 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3gpc: Crystal structure of human Acyl-CoA synthetase medium-chain famil... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3gpc | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human Acyl-CoA synthetase medium-chain family member 2A (L64P mutation) in a complex with CoA | |||||||||
 Components Components | Acyl-coenzyme A synthetase ACSM2A | |||||||||
 Keywords Keywords | LIGASE / MIDDLE-CHAIN ACYL-COA SYNTHETASE / XENOBIOTIC/MEDIUM-CHAIN FATTY ACID-COA LIGASE / ATP-BINDING / FATTY ACID METABOLISM / LIPID METABOLISM / MAGNESIUM / METAL-BINDING / MITOCHONDRION / NUCLEOTIDE-BINDING POLYMORPHISM / TRANSIT PEPTIDE / NUCLEOTIDE-BINDING | |||||||||
| Function / homology |  Function and homology information Function and homology informationdecanoate-CoA ligase activity / Conjugation of salicylate with glycine / medium-chain acyl-CoA ligase / fatty acid ligase activity / benzoate-CoA ligase / benzoate-CoA ligase activity / medium-chain fatty-acyl-CoA metabolic process / medium-chain fatty acid-CoA ligase activity / fatty-acyl-CoA synthase activity / acyl-CoA metabolic process ...decanoate-CoA ligase activity / Conjugation of salicylate with glycine / medium-chain acyl-CoA ligase / fatty acid ligase activity / benzoate-CoA ligase / benzoate-CoA ligase activity / medium-chain fatty-acyl-CoA metabolic process / medium-chain fatty acid-CoA ligase activity / fatty-acyl-CoA synthase activity / acyl-CoA metabolic process / triglyceride homeostasis / Aspirin ADME / fatty acid biosynthetic process / glucose homeostasis / mitochondrial matrix / mitochondrion / ATP binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Pilka, E.S. / Kochan, G.T. / Yue, W.W. / Bhatia, C. / Von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Weigelt, J. / Bountra, C. / Oppermann, U. / Structural Genomics Consortium (SGC) | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2009 Title: Structural snapshots for the conformation-dependent catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A Authors: Kochan, G. / Pilka, E.S. / von Delft, F. / Oppermann, U. / Yue, W.W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3gpc.cif.gz | 242 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3gpc.ent.gz | 189.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3gpc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3gpc_validation.pdf.gz | 1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3gpc_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 3gpc_validation.xml.gz | 55.4 KB | Display | |
| Data in CIF | 3gpc_validation.cif.gz | 76.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gp/3gpcftp://data.pdbj.org/pub/pdb/validation_reports/gp/3gpc | HTTPS FTP |
-Related structure data
| Related structure data |  2vzeC  2wd9C  3b7wSC  3c5eC  3dayC  3eq6C C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 63334.562 Da / Num. of mol.: 2 / Fragment: UNP residues 32-577 / Mutation: L64P Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ACSM2, ACSM2A, MACS2 / Production host:  TRICHOPLUSIA NI (cabbage looper) / Strain (production host): HIGH FIVE / References: UniProt: Q08AH3, medium-chain acyl-CoA ligase TRICHOPLUSIA NI (cabbage looper) / Strain (production host): HIGH FIVE / References: UniProt: Q08AH3, medium-chain acyl-CoA ligase#2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 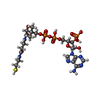  Mass: 767.534 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H36N7O16P3S Mass: 767.534 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H36N7O16P3S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 784 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 784 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | MUCH OF THE COA PANTETHEIN | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.53 Å3/Da / Density % sol: 51.39 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 25% PEG3350, 0.20M (NH4)2SO4, 0.1M BIS-TRIS, pH5.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1.5418 Å / Beamline: X10SA / Wavelength: 1.5418 Å |
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: May 10, 2008 |
| Radiation | Monochromator: OSMIC / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→50 Å / Num. all: 77119 / Num. obs: 73279 / % possible obs: 77.9 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 2.4 % / Rmerge(I) obs: 0.074 / Rsym value: 0.053 / Net I/σ(I): 8.3 |
| Reflection shell | Resolution: 1.9→2 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.355 / Mean I/σ(I) obs: 2.5 / Num. unique all: 11811 / Rsym value: 0.265 / % possible all: 81.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3B7W Resolution: 1.9→50 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.923 / SU B: 6.797 / SU ML: 0.09 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.171 / ESU R Free: 0.168 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.683 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
