 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3a10: Crystal structure of response regulator protein TrrA (TM1360) fro... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3a10 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of response regulator protein TrrA (TM1360) from Thermotoga maritima in complex with Mg(2+)-BeF (SeMet, L89M) | ||||||
 Components Components | Response regulator | ||||||
 Keywords Keywords | SIGNALING PROTEIN / phosphoacceptor | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphorelay signal transduction system / transcription cis-regulatory region binding / regulation of DNA-templated transcription / DNA-templated transcription / metal ion binding Similarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.63 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.63 Å | ||||||
 Authors Authors | Yamada, S. / Sugimoto, H. / Kobayashi, M. / Ohno, A. / Nakamura, H. / Shiro, Y. | ||||||
 Citation Citation | Journal: Structure / Year: 2009 Title: Structure of PAS-linked histidine kinase and the response regulator complex Authors: Yamada, S. / Sugimoto, H. / Kobayashi, M. / Ohno, A. / Nakamura, H. / Shiro, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3a10.cif.gz | 41.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3a10.ent.gz | 28.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3a10.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a1/3a10ftp://data.pdbj.org/pub/pdb/validation_reports/a1/3a10 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3a0rC  3a0sC  3a0tC  3a0uC  3a0vC  3a0wC  3a0xC  3a0yC  3a0zC C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13520.953 Da / Num. of mol.: 1 / Mutation: L89MSE Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (bacteria) / Gene: TM_1360 / Plasmid: pRSETA / Production host: | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||||
| #3: Chemical | ChemComp-BEF / 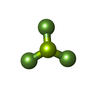  Mass: 66.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BeF3 Mass: 66.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BeF3 | ||||
| #4: Chemical |   Mass: 194.226 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 37.47 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 6.2 Details: 50% PEG200, 0.1M sodium/potassium-phosphate, 0.1M sodium chloride, pH 6.2, VAPOR DIFFUSION, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL45XU / Wavelength: 0.9795,0.9797,0.9787 / Beamline: BL45XU / Wavelength: 0.9795,0.9797,0.9787 | ||||||||||||
| Detector | Type: RIGAKU JUPITER 210 / Detector: CCD / Date: Mar 14, 2006 | ||||||||||||
| Radiation | Monochromator: DIAMOND 111 DOUBLE CRYSTAL MONOCHROMATOR / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 1.63→50 Å / Num. obs: 14298 / % possible obs: 98.9 % / Observed criterion σ(I): -3 / Redundancy: 7.3 % / Biso Wilson estimate: 18.8 Å2 / Rsym value: 0.037 / Net I/σ(I): 43.7 | ||||||||||||
| Reflection shell | Resolution: 1.63→1.69 Å / Redundancy: 6.4 % / Mean I/σ(I) obs: 8.9 / Rsym value: 0.143 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.63→20 Å / Cor.coef. Fo:Fc: 0.951 / Cor.coef. Fo:Fc free: 0.929 / Occupancy max: 1 / Occupancy min: 0.2 / SU B: 2.014 / SU ML: 0.071 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.116 / ESU R Free: 0.119 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 49.85 Å2 / Biso mean: 19.765 Å2 / Biso min: 8.65 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.63→20 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.631→1.673 Å / Total num. of bins used: 20
|
