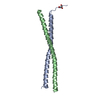
 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 357d | ||||||
|---|---|---|---|---|---|---|---|
| Title | 3.5 A structure of fragment I from E. coli 5S RRNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA / U-RNA / DOUBLE HELIX / KINKED / INTERNAL LOOP / OVERHANGING BASE / MODIFIED / MISMATCHED | ||||||
| Function / homology | : / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD PHASES FROM A BROMINE WERE COMBINED WITH MAD, SIRAS PHASES FROM A MERCURY DERIVIATIVE. MLPHARE WAS USED TO CALCULATE THE PHASES WHILE SIGMAA WAS USED TO COMBINE THE THREE PHASES SETS. / Resolution: 3.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD PHASES FROM A BROMINE WERE COMBINED WITH MAD, SIRAS PHASES FROM A MERCURY DERIVIATIVE. MLPHARE WAS USED TO CALCULATE THE PHASES WHILE SIGMAA WAS USED TO COMBINE THE THREE PHASES SETS. / Resolution: 3.5 Å | ||||||
 Authors Authors | Correll, C.C. / Freeborn, B. / Moore, P.B. / Steitz, T.A. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1997 Title: Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Authors: Correll, C.C. / Freeborn, B. / Moore, P.B. / Steitz, T.A. #1: Journal: J.Biomol.Struct.Dyn. / Year: 1997Title: Use of Chemically Modified Nucleotides to Determine a 62-Nucleotide RNA Crystal Structure: A Survey of Phosphorothioates, Br, Pt, and Hg Authors: Correll, C.C. / Freeborn, B. / Moore, P.B. / Steitz, T.A. #2: Journal: To be PublishedTitle: The Solution Structure of the Loop E/Loop D Region of E. Coli 5S rRNA Authors: Dallas, A. / Moore, P.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 357d.cif.gz | 45.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb357d.ent.gz | 31.9 KB | Display | PDB format |
| PDBx/mmJSON format | 357d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/57/357dftp://data.pdbj.org/pub/pdb/validation_reports/57/357d | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj










































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 3514.196 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||
|---|---|---|---|
| #2: RNA chain | Mass: 6166.682 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||
| #3: RNA chain | Mass: 10012.047 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||
| #4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg#5: Chemical | ChemComp-HG / |   Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 59 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion / pH: 6.4 / Details: pH 6.40, VAPOR DIFFUSION | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Beamline: X4A |
| Detector | Type: FUJI / Detector: IMAGE PLATE / Date: Apr 30, 1996 / Details: SILICON 111 BENDING MIRROR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 3.5→20 Å / Num. obs: 6037 / % possible obs: 99.1 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 4.9 % / Biso Wilson estimate: 56.6 Å2 / Rmerge(I) obs: 0.057 / Rsym value: 0.057 / Net I/σ(I): 11.3 |
| Reflection shell | Resolution: 3.5→3.62 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.291 / Mean I/σ(I) obs: 3.7 / Rsym value: 0.291 / % possible all: 95.1 |
| Reflection | *PLUS Highest resolution: 3.5 Å / Lowest resolution: 10 Å / Num. obs: 3602 / % possible obs: 99.5 % / Redundancy: 4.9 % / Num. measured all: 29667 |
| Reflection shell | *PLUS Highest resolution: 3.5 Å / Lowest resolution: 3.62 Å / Redundancy: 4.5 % / Mean I/σ(I) obs: 3.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD PHASES FROM A BROMINE WERE COMBINED WITH MAD, SIRAS PHASES FROM A MERCURY DERIVIATIVE. MLPHARE WAS USED TO CALCULATE THE PHASES WHILE SIGMAA WAS USED TO COMBINE THE THREE PHASES SETS. Resolution: 3.5→10 Å / Rfactor Rfree error: 0.017 / Data cutoff high absF: 18352194.5 / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0 Details: TARGET FOR REFINEMENT WAS MLHL: MAXIMUM LIKELIHOOD TARGET USING AMPLITUDES AND PHASE PROBABILITY DISTRIBUTION FROM THE COMBINED PHASES DESCRIBED BELOW.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 98.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.5→3.71 Å / Rfactor Rfree error: 0.05 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3.5 Å / Lowest resolution: 10 Å / σ(F): 0 / % reflection Rfree: 11 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 98.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 3.5 Å / % reflection Rfree: 13.2 % |
