 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ymt: gamma 2 adaptin EAR domain crystal structure with phage peptide G... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ymt | ||||||
|---|---|---|---|---|---|---|---|
| Title | gamma 2 adaptin EAR domain crystal structure with phage peptide GEEWGPWV | ||||||
 Components Components |
| ||||||
 Keywords Keywords | PROTEIN TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationAP-1 adaptor complex / Golgi to vacuole transport / clathrin-cargo adaptor activity / Golgi-associated vesicle / Lysosome Vesicle Biogenesis / transport vesicle / vesicle-mediated transport / intracellular protein transport / endosome membrane / Golgi membrane ...AP-1 adaptor complex / Golgi to vacuole transport / clathrin-cargo adaptor activity / Golgi-associated vesicle / Lysosome Vesicle Biogenesis / transport vesicle / vesicle-mediated transport / intracellular protein transport / endosome membrane / Golgi membrane / synapse / Golgi apparatus / membrane Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human)SYNTHETIC CONSTRUCT (others) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.802 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.802 Å | ||||||
 Authors Authors | Juergens, M.C. / Voros, J. / Rautureau, G. / Shepherd, D. / Pye, V.E. / Muldoon, J. / Johnson, C.M. / Ashcroft, A. / Freund, S.M.V. / Ferguson, N. | ||||||
 Citation Citation | Journal: Nat.Chem.Biol. / Year: 2013 Title: The Hepatitis B Virus Pres1 Domain Hijacks Host Trafficking Proteins by Motif Mimicry. Authors: Jurgens, M.C. / Voros, J. / Rautureau, G.J.P. / Shepherd, D.A. / Pye, V.E. / Muldoon, J. / Johnson, C.M. / Ashcroft, A.E. / Freund, S.M.V. / Ferguson, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ymt.cif.gz | 71.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ymt.ent.gz | 52.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2ymt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ym/2ymtftp://data.pdbj.org/pub/pdb/validation_reports/ym/2ymt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3zhfC  4bcxSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13762.746 Da / Num. of mol.: 1 / Fragment: EAR DOMAIN, RESIDUES 665-785 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  |
|---|---|
| #2: Protein/peptide | Mass: 957.019 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) SYNTHETIC CONSTRUCT (others) |
| #3: Chemical | ChemComp-PDO / 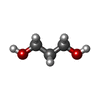  Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.53 Å3/Da / Density % sol: 51.42 % |
|---|---|
| Crystal grow | pH: 6.5 Details: 100 MM MES/IMIDAZOLE PH 6.5, 0.02 M 1,6-HEXANEDIOL, 0.02 M 1-BUTANOL, 0.02 M (RS)-1,2- PROPANEDIOL, 0.02 M 2-PROPANOL, 0.02 M 1,4-BUTANEDIOL, 0.02 M 1,3-PROPANEDIOL, 10% (W/V) PEG 20000 AND ...Details: 100 MM MES/IMIDAZOLE PH 6.5, 0.02 M 1,6-HEXANEDIOL, 0.02 M 1-BUTANOL, 0.02 M (RS)-1,2- PROPANEDIOL, 0.02 M 2-PROPANOL, 0.02 M 1,4-BUTANEDIOL, 0.02 M 1,3-PROPANEDIOL, 10% (W/V) PEG 20000 AND 24% (V/V) PEG400 (USING A 2:1 RATIO OF PROTEIN TO MOTHER LIQUOR) |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU IMAGE PLATE / Detector: IMAGE PLATE / Date: Sep 25, 2012 / Details: OSMIC BLUE CONFOCAL MIRRORS |
| Radiation | Monochromator: OSMIC VARIMAX HR OPTICS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→30.71 Å / Num. obs: 13212 / % possible obs: 97.7 % / Observed criterion σ(I): 2.7 / Redundancy: 6.4 % / Biso Wilson estimate: 17.2 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 11.6 |
| Reflection shell | Resolution: 1.8→1.9 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.49 / Mean I/σ(I) obs: 2.7 / % possible all: 8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4BCX Resolution: 1.802→30.708 Å / SU ML: 0.19 / σ(F): 1.34 / Phase error: 22.19 / Stereochemistry target values: ML Details: SAME KSOL AND BSOL ISSUE AS FOR THE 4BCX ENTRY. THEY DO NOT APPEAR IN THE SOFTWARE OUTPUT. PDB COMMENT WAS THAT THIS IS KNOWN AND NULL IS OK TO PUT INSTEAD OF N A AS THIS IS NOT ACCEPTED DURING DEPOSITION.
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 0 Å2 / ksol: 0 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.802→30.708 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -2.5745 Å / Origin y: -3.2126 Å / Origin z: 14.8178 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: ALL |
