 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2y90: Crystal structure of Hfq riboregulator from E. coli (P6 space group) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2y90 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Hfq riboregulator from E. coli (P6 space group) | ||||||
 Components Components | PROTEIN HFQ | ||||||
 Keywords Keywords | RNA BINDING PROTEIN / RNA-BINDING PROTEIN / SM-LIKE / RNA CHAPERONE | ||||||
| Function / homology |  Function and homology information Function and homology informationsRNA-mediated post-transcriptional gene silencing / positive regulation of translation, ncRNA-mediated / bacterial nucleoid / regulation of translation, ncRNA-mediated / bent DNA binding / regulation of RNA stability / RNA folding chaperone / tRNA processing / tRNA binding / regulation of DNA-templated transcription ...sRNA-mediated post-transcriptional gene silencing / positive regulation of translation, ncRNA-mediated / bacterial nucleoid / regulation of translation, ncRNA-mediated / bent DNA binding / regulation of RNA stability / RNA folding chaperone / tRNA processing / tRNA binding / regulation of DNA-templated transcription / DNA binding / RNA binding / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.252 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.252 Å | ||||||
 Authors Authors | Basquin, J. / Sauter, C. | ||||||
 Citation Citation | Journal: Cryst. Growth Des. / Year: 2011 Title: Exploiting Protein Engineering and Crystal Polymorphism for Successful X-Ray Structure Determination Authors: Bonnefond, L. / Schellenberger, P. / Basquin, J. / Demangeat, G. / Ritzenthaler, C. / Chenevert, R. / Balg, C. / Frugier, M. / Rudinger-Thirion, J. / Giege, R. / Lorber, L. / Sauter, C. #1: Journal: Nucleic Acids Res. / Year: 2003Title: Sm-Like Proteins in Eubacteria: The Crystal Structure of the Hfq Protein from Escherichia Coli. Authors: Sauter, C. / Basquin, J. / Suck, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2y90.cif.gz | 26 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2y90.ent.gz | 15.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2y90.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y9/2y90ftp://data.pdbj.org/pub/pdb/validation_reports/y9/2y90 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2yhtC  3zxiC  4v5wC  1hk9S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj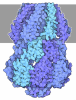

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 6
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11307.484 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | TWO AMINO ACIDS ADDED IN N-TERMINUS AND PROBABLE PROTEOLYTIC DEGRADATION AFTER RESIDUE 69. RESIDUES ...TWO AMINO ACIDS ADDED IN N-TERMINUS AND PROBABLE PROTEOLYTI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.7 Å3/Da / Density % sol: 30 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop Details: CRYSTALS WERE OBTAINED BY VAPOR DIFFUSION IN 2UL HANGING DROPS AT 20C. THE RESERVOIR CONTAINED 1.6 M AMMONIUM SULFATE, 0.1 M TRIS-HCL PH 8.0. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection twin | Operator: h,-h-k,-l / Fraction: 0.313 |
| Reflection | Resolution: 2.25→54 Å / Num. obs: 2979 / % possible obs: 99 % / Observed criterion σ(I): -3 / Redundancy: 7.6 % / Biso Wilson estimate: 28.6 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 11.7 |
| Reflection shell | Resolution: 2.25→2.3 Å / Rmerge(I) obs: 0.17 / Mean I/σ(I) obs: 4.3 / % possible all: 89.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HK9 Resolution: 2.252→53.261 Å / σ(F): 0 / Phase error: 32.43 / Stereochemistry target values: TWIN_LSQ_F Details: THE FULL LENGTH HFQ PROTEIN CRYSTALLIZED AFTER PROTEOLYTIC DEGRADATION AS INDICATED BY THE CRYSTAL SOLVENT SOLVENT CONTENT (SEE REF1). THE RESULTING MONOMER LACKS RESIDUES 70-102. THE LATTER ...Details: THE FULL LENGTH HFQ PROTEIN CRYSTALLIZED AFTER PROTEOLYTIC DEGRADATION AS INDICATED BY THE CRYSTAL SOLVENT SOLVENT CONTENT (SEE REF1). THE RESULTING MONOMER LACKS RESIDUES 70-102. THE LATTER ARE EITHER DISORDERED IN THE CRYSTAL OR ABSENT DUE TO BY PROTEOLYSIS.
| ||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.72 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 25.035 Å2 / ksol: 0.333 e/Å3 | ||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.9 Å2
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.252→53.261 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2608→24.9582 Å
|
