 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2y65: Crystal structure of Drosophila melanogaster kinesin-1 motor doma... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2y65 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Drosophila melanogaster kinesin-1 motor domain dimer-tail complex | ||||||
 Components Components | (KINESIN HEAVY CHAIN) x 2 | ||||||
 Keywords Keywords | MOTOR PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationactin filament bundle organization / ovarian nurse cell to oocyte transport / anterograde axonal transport of mitochondrion / mitochondrion distribution / regulation of pole plasm oskar mRNA localization / anterograde dendritic transport / eye photoreceptor cell differentiation / oocyte dorsal/ventral axis specification / larval locomotory behavior / pole plasm assembly ...actin filament bundle organization / ovarian nurse cell to oocyte transport / anterograde axonal transport of mitochondrion / mitochondrion distribution / regulation of pole plasm oskar mRNA localization / anterograde dendritic transport / eye photoreceptor cell differentiation / oocyte dorsal/ventral axis specification / larval locomotory behavior / pole plasm assembly / dorsal appendage formation / COPI-dependent Golgi-to-ER retrograde traffic / Kinesins / oocyte microtubule cytoskeleton polarization / pole plasm oskar mRNA localization / larval somatic muscle development / centrosome separation / anterograde dendritic transport of neurotransmitter receptor complex / microtubule sliding / transport along microtubule / actin cap / microtubule plus-end / plus-end-directed microtubule motor activity / axo-dendritic transport / microtubule motor activity / kinesin complex / microtubule-based movement / nuclear migration / dendrite morphogenesis / stress granule disassembly / intracellular distribution of mitochondria / tropomyosin binding / synaptic vesicle transport / microtubule polymerization / cytoskeletal motor activity / axon cytoplasm / axonogenesis / dendrite cytoplasm / axon guidance / microtubule binding / microtubule / ATP hydrolysis activity / ATP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Kaan, H.Y.K. / Hackney, D.D. / Kozielski, F. | ||||||
 Citation Citation | Journal: Science / Year: 2011 Title: The Structure of the Kinesin-1 Motor-Tail Complex Reveals the Mechanism of Autoinhibition. Authors: Kaan, H.Y.K. / Hackney, D.D. / Kozielski, F. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2y65.cif.gz | 301.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2y65.ent.gz | 243.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2y65.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y6/2y65ftp://data.pdbj.org/pub/pdb/validation_reports/y6/2y65 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2y5wSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 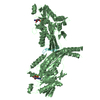
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.9805, 0.0458, -0.1911), Vector: Details | OWING TO ITS AMINO ACID SEQUENCE, CHAINS X AND Y ARE ALMOST SYMMETRICAL, IN TERMS OF SIDE CHAIN PROPERTIES, ABOUT THE LYS 944 RESIDUE. THIS DISTINCTIVE FEATURE OF THE CHAINS X AND Y ALLOWS THEM TO BIND IN TWO DIRECTIONS BETWEEN THE DIMERS A-A' AND B-B', WHICH ALSO EXHIBIT TWO-FOLD SYMMETRY. | |
-Components
| #1: Protein | Mass: 40885.301 Da / Num. of mol.: 4 / Fragment: MOTOR DOMAIN, RESIDUES 1-365 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Protein/peptide | Mass: 1913.098 Da / Num. of mol.: 3 / Fragment: TAIL DOMAIN RESIDUES 937-952 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 656 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 656 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.87 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.4 Details: 18% POLYETHYLENE GLYCOL-3350, 0.2M POTASSIUM CHLORIDE, 0.1M HEPES SODIUM PH 7.4 |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 / Beamline: X06SA / Wavelength: 1 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 16, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 80517 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 8.8 % / Biso Wilson estimate: 41.383 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 21.7 |
| Reflection shell | Resolution: 2.2→2.32 Å / Redundancy: 8.3 % / Rmerge(I) obs: 0.37 / Mean I/σ(I) obs: 5.5 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2Y5W Resolution: 2.2→50 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.924 / SU B: 5.611 / SU ML: 0.147 / Cross valid method: THROUGHOUT / ESU R: 0.276 / ESU R Free: 0.221 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. PSEUDOSYMMETRY IN THE CRYSTAL STRUCTURE, WHERE CHAINS X AND Y LIE ON THE TWO-FOLD CRYSTALLOGRAPHIC SYMMETRY AXIS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.405 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|