 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2wys: High resolution crystallographic structure of the Clostridium the... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wys | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | High resolution crystallographic structure of the Clostridium thermocellum N-terminal endo-1,4-beta-D-xylanase 10B (Xyn10B) CBM22-1- GH10 modules complexed with xylohexaose | |||||||||
 Components Components | ENDO-1,4-BETA-XYLANASE Y | |||||||||
 Keywords Keywords | HYDROLASE / XYLAN DEGRADATION / CELLULOSOME / GLYCOSIDASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationcellulosome / endo-1,4-beta-xylanase / endo-1,4-beta-xylanase activity / xylan catabolic process Similarity search - Function | |||||||||
| Biological species |  CLOSTRIDIUM THERMOCELLUM (bacteria) CLOSTRIDIUM THERMOCELLUM (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.75 Å | |||||||||
 Authors Authors | Najmudin, S. / Pinheiro, B.A. / Romao, M.J. / Prates, J.A.M. / Fontes, C.M.G.A. | |||||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 2010 Title: Putting an N-Terminal End to the Clostridium Thermocellum Xylanase Xyn10B Story: Crystal Structure of the Cbm22-1-Gh10 Modules Complexed with Xylohexaose. Authors: Najmudin, S. / Pinheiro, B.A. / Prates, J.A.M. / Gilbert, H.J. / Romao, M.J. / Fontes, C.M.G.A. #1: Journal: Acta Crystallogr.,Sect.F / Year: 2008 Title: Purification, Crystallization and Crystallographic Analysis of Clostridium Thermocellum Endo-1,4-Beta- D-Xylanase 10B in Complex with Xylohexaose. Authors: Najmudin, S. / Pinheiro, B.A. / Romao, M.J. / Prates, J.A.M. / Fontes, C.M.G.A. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AF" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AF" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BE" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wys.cif.gz | 226.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wys.ent.gz | 179.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2wys.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wy/2wysftp://data.pdbj.org/pub/pdb/validation_reports/wy/2wys | HTTPS FTP |
|---|
-Related structure data






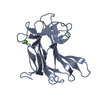















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 60658.254 Da / Num. of mol.: 2 / Fragment: CBM22-1, GH10, RESIDUES 33-551 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) CLOSTRIDIUM THERMOCELLUM (bacteria) / Strain: YS / Plasmid: PGEM T-EASY, PET21A / Production host: |
|---|
-Sugars , 3 types, 3 molecules 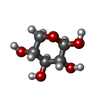
| #2: Polysaccharide | beta-D-xylopyranose-(1-2)-beta-D-xylopyranose Source method: isolated from a genetically manipulated source |
|---|---|
| #3: Polysaccharide | beta-D-xylopyranose-(1-4)-beta-D-xylopyranose-(1-4)-beta-D-xylopyranose / 4beta-beta-xylotriose  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: 4beta-beta-xylotriose |
| #4: Sugar | ChemComp-XYP /  Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C5H10O5 |
-Non-polymers , 3 types, 384 molecules 


| #5: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#6: Chemical |  Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 378 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | ENGINEERED| Has protein modification | Y | Nonpolymer details | POTASSIUM (K): FROM THE CRYSTALLIZATION BUFFER PHOSPHATE ION (PO4): FRO THE CRYSTALLIZATION BUFFER ...POTASSIUM (K): FROM THE CRYSTALLIZ | Sequence details | THE PROTEIN SEQUENCE OF THE CONSTRUCT CORRESPONDS TO AMINO ACID RESIDUES 32 TO 551 WITH THE E337A ...THE PROTEIN SEQUENCE OF THE CONSTRUCT CORRESPOND | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.9 Å3/Da / Density % sol: 75 % Description: PEAK DATA WAS THE BEST DATA COLLECTED OVER THE THREE WAVELENGTHS AND WAS USED FOR MODELING THE STRUCTURE |
|---|---|
| Crystal grow | Temperature: 292 K / Details: 1M KH2PO4 AT 292K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9791, 0.9793, 0.9756 / Beamline: ID29 / Wavelength: 0.9791, 0.9793, 0.9756 | ||||||||||||
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jul 22, 2007 | ||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.75→100.5 Å / Num. obs: 61428 / % possible obs: 100 % / Observed criterion σ(I): 1.4 / Redundancy: 9.6 % / Biso Wilson estimate: 71.1 Å2 / Rmerge(I) obs: 0.19 / Net I/σ(I): 13 | ||||||||||||
| Reflection shell | Resolution: 2.75→2.8 Å / Redundancy: 9.1 % / Rmerge(I) obs: 1 / Mean I/σ(I) obs: 1.4 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD Starting model: NONE Resolution: 2.75→86.85 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.921 / SU B: 10.849 / SU ML: 0.205 / Cross valid method: THROUGHOUT / ESU R: 0.309 / ESU R Free: 0.253 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 66.377 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→86.85 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|