 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vop: Crystal structure of N-terminal domains of Human La protein compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vop | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of N-terminal domains of Human La protein complexed with RNA oligomer AUUUU | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA BINDING PROTEIN / RNA-BINDING PROTEIN / RNA RECOGNITION MOTIF / SYSTEMIC LUPUS ERYTHEMATOSUS / PHOSPHOPROTEIN / RNA MATURATION / NUCLEUS / LA MOTIF / RNA-BINDING / POLYMORPHISM | ||||||
| Function / homology |  Function and homology information Function and homology informationnuclear histone mRNA catabolic process / histone mRNA metabolic process / tRNA 3'-end processing / protein localization to cytoplasmic stress granule / IRES-dependent viral translational initiation / RNA Polymerase III Transcription Termination / tRNA modification / tRNA export from nucleus / RNA Polymerase III Abortive And Retractive Initiation / sequence-specific mRNA binding ...nuclear histone mRNA catabolic process / histone mRNA metabolic process / tRNA 3'-end processing / protein localization to cytoplasmic stress granule / IRES-dependent viral translational initiation / RNA Polymerase III Transcription Termination / tRNA modification / tRNA export from nucleus / RNA Polymerase III Abortive And Retractive Initiation / sequence-specific mRNA binding / tRNA 5'-leader removal / tRNA processing / poly(U) RNA binding / positive regulation of translation / cytoplasmic stress granule / chromosome, telomeric region / tRNA binding / ribonucleoprotein complex / mRNA binding / RNA binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Kotik-Kogan, O. / Valentine, E.R. / Sanfelice, D. / Conte, M.R. / Curry, S. | ||||||
 Citation Citation | Journal: Structure / Year: 2008 Title: Structural Analysis Reveals Conformational Plasticity in the Recognition of RNA 3' Ends by the Human La Protein. Authors: Kotik-Kogan, O. / Valentine, E.R. / Sanfelice, D. / Conte, M.R. / Curry, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vop.cif.gz | 53.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vop.ent.gz | 36.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2vop.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vo/2vopftp://data.pdbj.org/pub/pdb/validation_reports/vo/2vop | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2vodC  2vonC  2vooC  1zh5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 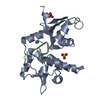
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22529.809 Da / Num. of mol.: 1 / Fragment: N-TERMINAL DOMAIN, RESIDUES 4-194 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PETM11 / Production host:  | ||
|---|---|---|---|
| #2: RNA chain | Mass: 1508.912 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) | ||
| #3: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4Sequence details | FIRST TWO RESIDUES ARE FROM VECTOR (GS) REMAINING SEQUENCE CORRESPOND | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.15 Å3/Da / Density % sol: 70.16 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 0.62 / Beamline: ID23-1 / Wavelength: 0.62 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jul 12, 2006 / Details: SINGLE SILICON (111) MONOCHROMATOR |
| Radiation | Monochromator: SINGLE SILICON (111) MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.62 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→63 Å / Num. obs: 8894 / % possible obs: 98.3 % / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Biso Wilson estimate: 76.5 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 2.8→2.95 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.37 / Mean I/σ(I) obs: 1.9 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ZH5 Resolution: 2.8→48.24 Å / Rfactor Rfree error: 0.013 / Data cutoff high absF: 1603544.48 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→48.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.98 Å / Total num. of bins used: 6
|
