 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2qbl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of ferric G248T cytochrome P450cam | ||||||
 Components Components | Cytochrome P450-cam | ||||||
 Keywords Keywords | OXIDOREDUCTASE / CYP101 / mutant / conserved active site residue / Gly248 / heme geometry | ||||||
| Function / homology |  Function and homology information Function and homology informationcamphor 5-monooxygenase / camphor 5-monooxygenase activity / (+)-camphor catabolic process / iron ion binding / heme binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.8 Å | ||||||
 Authors Authors | von Koenig, K. / Makris, T.M. / Sligar, S.D. / Schlichting, I. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2007 Title: Alteration of P450 Distal Pocket Solvent Leads to Impaired Proton Delivery and Changes in Heme Geometry. Authors: Makris, T.M. / Koenig, K.V. / Schlichting, I. / Sligar, S.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2qbl.cif.gz | 104.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2qbl.ent.gz | 77 KB | Display | PDB format |
| PDBx/mmJSON format | 2qbl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2qbl_validation.pdf.gz | 825.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2qbl_full_validation.pdf.gz | 831.5 KB | Display | |
| Data in XML | 2qbl_validation.xml.gz | 20.8 KB | Display | |
| Data in CIF | 2qbl_validation.cif.gz | 31.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qb/2qblftp://data.pdbj.org/pub/pdb/validation_reports/qb/2qbl | HTTPS FTP |
-Related structure data
| Related structure data |  2qbmC  2qbnC  2qboC  1akdS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 47592.996 Da / Num. of mol.: 1 / Mutation: G248T Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas putida (bacteria) / Gene: camC, cyp101 / Plasmid: pET28 / Species (production host): Escherichia coli / Production host: |
|---|---|
| #2: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K |
| #3: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 |
| #4: Chemical | ChemComp-CAM / 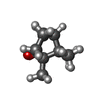  Mass: 152.233 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16O Mass: 152.233 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16O |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 371 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 371 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.73 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.4 Details: 1 UL OF 15 MG/ML P450 IN 50 MM Potassium phosphate, 250 MM KCL WERE MIXED WITH AN EQUAL VOLUME OF THE RESERVOIR SOLUTION (100 mM Tris, 250 mM KCl, 27-30% PEG 8000, 100 MM DTE), pH 7.4, VAPOR ...Details: 1 UL OF 15 MG/ML P450 IN 50 MM Potassium phosphate, 250 MM KCL WERE MIXED WITH AN EQUAL VOLUME OF THE RESERVOIR SOLUTION (100 mM Tris, 250 mM KCl, 27-30% PEG 8000, 100 MM DTE), pH 7.4, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction |
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.93 Å / Beamline: ID14-2 / Wavelength: 0.93 Å | |||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jul 23, 2004 / Details: toroidal mirror | |||||||||
| Radiation | Monochromator: diamond (111, Ge(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength | Wavelength: 0.93 Å / Relative weight: 1 | |||||||||
| Reflection | Resolution: 1.8→20 Å / Num. all: 41485 / Num. obs: 41485 / % possible obs: 99.7 % / Observed criterion σ(I): -3 / Redundancy: 4.4 % / Rsym value: 0.047 / Net I/σ(I): 19.5 | |||||||||
| Reflection shell | Resolution: 1.8→1.9 Å / Redundancy: 4.5 % / Mean I/σ(I) obs: 4.4 / Num. unique all: 6120 / Rsym value: 0.381 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB entry 1akd Resolution: 1.8→20 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: maximum likelihood
| ||||||||||||||||||||||||||||
| Solvent computation | Bsol: 46.849 Å2 | ||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.264 Å2
| ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→20 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| Xplor file |
|
