 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2puf: CRYSTAL STRUCTURE OF THE LACI FAMILY MEMBER, PURR, BOUND TO DNA: ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2puf | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE LACI FAMILY MEMBER, PURR, BOUND TO DNA: MINOR GROOVE BINDING BY ALPHA HELICES | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / COMPLEX (DNA-BINDING PROTEIN-DNA) / DNA-BINDING REGULATORY PROTEIN / EXTENDED COREPRESSOR SPECIFICITY / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationguanine binding / negative regulation of purine nucleotide biosynthetic process / purine nucleotide biosynthetic process / DNA-binding transcription repressor activity / transcription cis-regulatory region binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / protein homodimerization activity / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 3 Å X-RAY DIFFRACTION / Resolution: 3 Å | ||||||
 Authors Authors | Lu, F. / Schumacher, M.A. / Arvidson, D.N. / Haldimann, A. / Wanner, B.L. / Zalkin, H. / Brennan, R.G. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Structure-based redesign of corepressor specificity of the Escherichia coli purine repressor by substitution of residue 190. Authors: Lu, F. / Schumacher, M.A. / Arvidson, D.N. / Haldimann, A. / Wanner, B.L. / Zalkin, H. / Brennan, R.G. #1: Journal: Science / Year: 1994Title: Crystal Structure of LacI Member, PurR, Bound to DNA: Minor Groove Binding by Alpha Helices Authors: Schumacher, M.A. / Choi, K.Y. / Zalkin, H. / Brennan, R.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2puf.cif.gz | 81.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2puf.ent.gz | 56.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2puf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2puf_validation.pdf.gz | 390 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2puf_full_validation.pdf.gz | 419.2 KB | Display | |
| Data in XML | 2puf_validation.xml.gz | 12.1 KB | Display | |
| Data in CIF | 2puf_validation.cif.gz | 17.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pu/2pufftp://data.pdbj.org/pub/pdb/validation_reports/pu/2puf | HTTPS FTP |
-Related structure data

















-Links
 PDBj
PDBj








































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE COMPLEX LIES ON A CRYSTALLOGRAPHIC TWO-FOLD AXIS, AND ONLY ONE MONOMER-DNA HALF-SITE CONSTITUTES THE ASYMMETRIC UNIT. THE COORDINATES COMPRISE ONE REPRESSOR MONOMER AND ONE DNA STRAND FOR THE ENTIRE SITE. THE FULL COMPLEX CAN BE CONSTRUCTED BY GENERATING THE SECOND HALF USING THE CRYSTALLOGRAPHIC SYMMETRY OPERATION (X, -Y, -Z). |
-Components
| #1: DNA chain | Mass: 5202.384 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: Protein | Mass: 38062.531 Da / Num. of mol.: 1 / Mutation: R190Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #3: Chemical | ChemComp-GUN / 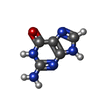  Mass: 151.126 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5O Mass: 151.126 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5O |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THE HYDROGEN BONDS FROM ARG 190 DETERMINE SPECIFICITY FOR THE O6 EXOCYCLIC ATOM OF THE NATURAL ...THE HYDROGEN BONDS FROM ARG 190 DETERMINE SPECIFICIT |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.94 Å3/Da / Density % sol: 68.79 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: pH 7.40, VAPOR DIFFUSION, HANGING DROP, temperature 293.00K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Details: Schumacher, M.A., (1994) J.Mol.Biol, 242, 302. / pH: 7.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: SDMS / Detector: AREA DETECTOR |
| Radiation | Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 3 Å / Num. obs: 13317 / % possible obs: 96 % / Redundancy: 3.75 % / Rmerge(I) obs: 0.098 / Net I/σ(I): 3.6 |
| Reflection | *PLUS Highest resolution: 3 Å / % possible obs: 96 % / Num. measured all: 49911 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3→10 Å / Isotropic thermal model: ISOTROPIC / σ(F): 1 / Stereochemistry target values: ENGH AND HUBER /
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: TNT / Bsol: 150 Å2 / ksol: 0.8 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3 Å / Lowest resolution: 10 Å / σ(F): 1 / Rfactor Rwork: 0.222 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
