 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1efa: CRYSTAL STRUCTURE OF THE LAC REPRESSOR DIMER BOUND TO OPERATOR AN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1efa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE LAC REPRESSOR DIMER BOUND TO OPERATOR AND THE ANTI-INDUCER ONPF | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSCRIPTION/DNA / protein-dna complex / helix-turn-helix / gene regulation / molecular switch / TRANSCRIPTION-DNA COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationDNA-binding transcription repressor activity / cis-regulatory region sequence-specific DNA binding / transcription cis-regulatory region binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.6 Å | |||||||||
 Authors Authors | Bell, C.E. / Lewis, M. | |||||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2000 Title: A closer view of the conformation of the Lac repressor bound to operator. Authors: Bell, C.E. / Lewis, M. #1: Journal: Science / Year: 1996Title: Crystal structure of the lactose operon repressor and its complexes with DNA and inducer Authors: Lewis, M. / Chang, G. / Horton, N.C. / Kercher, M.A. / Pace, H.C. #2: Journal: Science / Year: 1995Title: Crystal structure of lac repressor core tetramer and its implications for DNA looping Authors: Friedman, A.M. / Fischmann, T.O. / Steitz, T.A. #3: Journal: Structure / Year: 1999Title: The solution structure of lac repressor headpiece 62 complexed to a symmetrical lac operator sequence determined by NMR and restrained molecular dynamics Authors: Spronk, C.A.E.M. / Bonvin, A.M.J.J. / Radha, P.K. / Melacini, G. / Boelens, R. #4: Journal: J.Mol.Biol. / Year: 1996Title: Refined structure of lac repressor headpiece (1-56) determined by relaxation matrix calculations from 2D and 3D NOE data: change of tertiary structure upon binding to the lac operator Authors: Slijper, M. / Bonvin, A.M. / Boelens, R. / Kaptein, R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1efa.cif.gz | 205.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1efa.ent.gz | 161.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1efa.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ef/1efaftp://data.pdbj.org/pub/pdb/validation_reports/ef/1efa | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 6462.196 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: Protein | Mass: 35756.797 Da / Num. of mol.: 3 / Fragment: RESIDUES 1-333 / Mutation: ALA109THR Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Sugar | 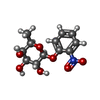  Type: D-saccharide / Mass: 285.250 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H15NO7 Type: D-saccharide / Mass: 285.250 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H15NO7#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: ammonium sulfate, PEG 400, HEPES, Glycerol, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 / Beamline: X25 / Wavelength: 1.1 |
| Detector | Type: BRANDEIS - B4 / Detector: CCD / Date: Jan 11, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→17 Å / Num. all: 75989 / Num. obs: 74896 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 5.3 % / Biso Wilson estimate: 43.8 Å2 / Rmerge(I) obs: 0.066 / Net I/σ(I): 13.5 |
| Reflection shell | Resolution: 2.6→2.68 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.402 / Num. unique all: 6272 / % possible all: 100 |
| Reflection | *PLUS Num. measured all: 392400 |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.6→10 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 953713.59 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): -3 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 70.17 Å2 / ksol: 0.477 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 69.2 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→10 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: CONSTR | ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.76 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
