 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2pl8 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | D(GTATACC) under hydrostatic pressure of 1.04 GPa | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / HIGH-PRESSURE | Function / homology | SPERMINE / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.65 Å  Authors AuthorsPrange, T. / Girard, E. / Fourme, R. / Kahn, R. |  CitationJournal: Nucleic Acids Res. / Year: 2007 CitationJournal: Nucleic Acids Res. / Year: 2007Title: Adaptation of the base-paired double-helix molecular architecture to extreme pressure. Authors: Girard, E. / Prange, T. / Dhaussy, A.C. / Migianu-Griffoni, E. / Lecouvey, M. / Chervin, J.C. / Mezouar, M. / Kahn, R. / Fourme, R. #1: Journal: Nature / Year: 1989 Title: Coexistence of A-and B-Form DNA in a Single Crystal Lattice Authors: Doucet, J. / Benoit, J.-P. / Cruse, W.B.T. / Prange, T. / Kennard, O. #2: Journal: J.Synchrotron Radia. / Year: 2001 Title: High-pressure protein crystallography (HPPX): Instrumentation, methodology and results on lysozyme crystals Authors: Fourme, R. / Kahn, R. / Mezouar, M. / Girard, E. / Horentrup, C. / Prange, T. / Ascone, I. #3: Journal: BIOCHEM.BIOPHYS.ACTA PROTEINS & PROTEOMICS / Year: 2006Title: High pressure macromolecular crystallography: The 140 MPa crystal structure at 2.3 A resolution of urate oxidase, A 135 KD tetrameric assembly Authors: Colloc'h, N. / Girard, E. / Dhaussy, A.C. / Kahn, R. / Ascone, I. / Mezouar, M. / Fourme, R. #4: Journal: J.Mol.Biol. / Year: 1987Title: Crystal structure of hen egg-white lysozyme at a hydrostatic pressure of 1000 atmospheres. Authors: Kundrot, C.E. / Richards, F.M. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2pl8.cif.gz | 21.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2pl8.ent.gz | 13 KB | Display | PDB format |
| PDBx/mmJSON format | 2pl8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pl/2pl8ftp://data.pdbj.org/pub/pdb/validation_reports/pl/2pl8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2pkvC  2pl4SC  2plbC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 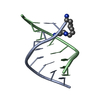
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 2426.617 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: synthetic DNA octamer #2: Chemical | ChemComp-SPM / | 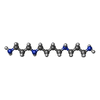  Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4 Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.63 % | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 20 mg of DNA dissolved in 0.2 ml of 15% MPD solution cacodylate buffer 10-2 M. Additives: 10-5 M sodium azide, 10-2 M MgCl2, 2.10-2 M spermine chloride. Reservoir: same solution but 50 % ...Details: 20 mg of DNA dissolved in 0.2 ml of 15% MPD solution cacodylate buffer 10-2 M. Additives: 10-5 M sodium azide, 10-2 M MgCl2, 2.10-2 M spermine chloride. Reservoir: same solution but 50 % MPD, pH 7, VAPOR DIFFUSION, SITTING DROP, temperature 290K | ||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID27 / Wavelength: 0.3738 Å / Beamline: ID27 / Wavelength: 0.3738 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Nov 10, 2006 / Details: 2 crystals, parallel beam |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.3738 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→18 Å / Num. all: 5079 / Num. obs: 5018 / % possible obs: 89.4 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 4 / Redundancy: 3.2 % / Rmerge(I) obs: 0.058 / Net I/σ(I): 6.6 |
| Reflection shell | Resolution: 1.65→1.75 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.251 / Mean I/σ(I) obs: 2.9 / Num. unique all: 733 / % possible all: 90.27 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 2PL4 Resolution: 1.65→10 Å / Num. parameters: 1671 / Num. restraintsaints: 1688 Isotropic thermal model: indiidual atom restrained B factors Cross valid method: FREE R / σ(F): 2 / σ(I): 4 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 184 / Occupancy sum non hydrogen: 406.8 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
