| 登録情報 | データベース: PDB / ID: 2o40
|
|---|
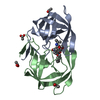
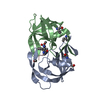
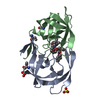
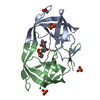
| タイトル | Crystal Structure of a Chemically Synthesized 203 Amino Acid 'Covalent Dimer' HIV-1 Protease Molecule |
|---|
 要素 要素 | covalent dimer HIV-1 protease |
|---|
 キーワード キーワード | Hydrolase/Hydrolase inhibitor / beta-turn / Hydrolase-Hydrolase inhibitor complex |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
aspartic-type endopeptidase activity / proteolysis類似検索 - 分子機能 Retropepsin-like catalytic domain / Retropepsins / Retroviral aspartyl protease / Aspartyl protease, retroviral-type family profile. / Peptidase A2A, retrovirus, catalytic / Cathepsin D, subunit A; domain 1 / Acid Proteases / Aspartic peptidase, active site / Eukaryotic and viral aspartyl proteases active site. / Aspartic peptidase domain superfamily ...Retropepsin-like catalytic domain / Retropepsins / Retroviral aspartyl protease / Aspartyl protease, retroviral-type family profile. / Peptidase A2A, retrovirus, catalytic / Cathepsin D, subunit A; domain 1 / Acid Proteases / Aspartic peptidase, active site / Eukaryotic and viral aspartyl proteases active site. / Aspartic peptidase domain superfamily / Beta Barrel / Mainly Beta類似検索 - ドメイン・相同性 N-{(2S)-2-[(N-acetyl-L-threonyl-L-isoleucyl)amino]hexyl}-L-norleucyl-L-glutaminyl-N~5~-[amino(iminio)methyl]-L-ornithinamide / Chem-2NC / Protease類似検索 - 構成要素 |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.65 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.65 Å |
|---|
 データ登録者 データ登録者 | Torbeev, V.Y. / Kent, S.B.H. |
|---|
 引用 引用 | ジャーナル: Angew.Chem.Int.Ed.Engl. / 年: 2007
タイトル: Convergent chemical synthesis and crystal structure of a 203 amino acid "covalent dimer" HIV-1 protease enzyme molecule.
著者: Torbeev, V.Y. / Kent, S.B. |
|---|
| 履歴 | | 登録 | 2006年12月2日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2006年12月19日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年5月1日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Advisory / Atomic model ...Advisory / Atomic model / Database references / Derived calculations / Non-polymer description / Structure summary / Version format compliance |
|---|
| 改定 1.3 | 2013年2月27日 | Group: Other |
|---|
| 改定 1.4 | 2017年10月18日 | Group: Refinement description / カテゴリ: software |
|---|
| 改定 1.5 | 2023年12月27日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_conn / struct_sheet / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_leaving_atom_flag / _struct_sheet.number_strands / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード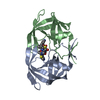















 PDBj
PDBj 集合体
集合体
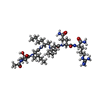
 タイプ: peptide-like, Peptide-like / クラス: 阻害剤 / 分子量: 770.983 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C35H68N11O8
タイプ: peptide-like, Peptide-like / クラス: 阻害剤 / 分子量: 770.983 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C35H68N11O8 分子量: 18.015 Da / 分子数: 57 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 57 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 23-ID-B / 波長: 0.97932 Å
/ ビームライン: 23-ID-B / 波長: 0.97932 Å 解析
解析