 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2j3s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human filamin A Ig domains 19 to 21 | ||||||
 Components Components | FILAMIN-A | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / CYTOSKELETON / PHOSPHORYLATION | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of membrane repolarization during atrial cardiac muscle cell action potential / regulation of membrane repolarization during cardiac muscle cell action potential / establishment of Sertoli cell barrier / formation of radial glial scaffolds / Myb complex / adenylate cyclase-inhibiting dopamine receptor signaling pathway / positive regulation of integrin-mediated signaling pathway / blood coagulation, intrinsic pathway / OAS antiviral response / protein localization to bicellular tight junction ...regulation of membrane repolarization during atrial cardiac muscle cell action potential / regulation of membrane repolarization during cardiac muscle cell action potential / establishment of Sertoli cell barrier / formation of radial glial scaffolds / Myb complex / adenylate cyclase-inhibiting dopamine receptor signaling pathway / positive regulation of integrin-mediated signaling pathway / blood coagulation, intrinsic pathway / OAS antiviral response / protein localization to bicellular tight junction / actin crosslink formation / positive regulation of actin filament bundle assembly / positive regulation of neuron migration / tubulin deacetylation / megakaryocyte development / Cell-extracellular matrix interactions / positive regulation of platelet activation / positive regulation of potassium ion transmembrane transport / apical dendrite / Fc-gamma receptor I complex binding / positive regulation of neural precursor cell proliferation / protein localization to cell surface / negative regulation of transcription by RNA polymerase I / podosome / wound healing, spreading of cells / GP1b-IX-V activation signalling / SMAD binding / receptor clustering / cortical cytoskeleton / RHO GTPases activate PAKs / semaphorin-plexin signaling pathway / mitotic spindle assembly / cilium assembly / potassium channel regulator activity / release of sequestered calcium ion into cytosol / positive regulation of substrate adhesion-dependent cell spreading / regulation of cell migration / protein localization to plasma membrane / dendritic shaft / actin filament / establishment of protein localization / negative regulation of protein catabolic process / protein sequestering activity / cerebral cortex development / positive regulation of protein import into nucleus / platelet aggregation / mRNA transcription by RNA polymerase II / G protein-coupled receptor binding / small GTPase binding / kinase binding / Z disc / cell-cell junction / actin filament binding / actin cytoskeleton / Platelet degranulation / growth cone / actin cytoskeleton organization / GTPase binding / DNA-binding transcription factor binding / perikaryon / transmembrane transporter binding / positive regulation of canonical NF-kappaB signal transduction / postsynapse / protein stabilization / cadherin binding / focal adhesion / nucleolus / negative regulation of apoptotic process / perinuclear region of cytoplasm / glutamatergic synapse / protein homodimerization activity / RNA binding / extracellular exosome / extracellular region / membrane / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Kiema, T.-R. / Ylanne, J. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2007 Title: Structure of Three Tandem Filamin Domains Reveals Auto-Inhibition of Ligand-Binding. Authors: Lad, Y. / Kiema, T.-R. / Jiang, P. / Pentikanen, O.T. / Coles, C.H. / Campbell, I.D. / Calderwood, D.A. / Ylanne, J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2j3s.cif.gz | 109.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2j3s.ent.gz | 82.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2j3s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j3/2j3sftp://data.pdbj.org/pub/pdb/validation_reports/j3/2j3s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1v05S S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Refine code: 1
NCS ensembles :
|
-Components
| #1: Protein | Mass: 30595.975 Da / Num. of mol.: 2 Fragment: IMMUNOGLOBULIN-LIKE DOMAINS 19 TO 21, RESIDUES 2045-2329 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  #2: Chemical |   Mass: 79.904 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Br Mass: 79.904 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Br#3: Chemical | ChemComp-DIO / 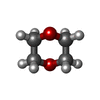  Mass: 88.105 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H8O2#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2OSequence details | THE THREE N-TERMINAL RESIDUES GLY ALA MET ORIGINATE FROM THE EXPRESSION | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.1 Details: PROTEIN WAS CRYSTALLIZED FROM 1.6M AMMONIUM SULPHATE, 0.1M CITRIC ACID PH 6.1, 10% DIOXANE. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 0.91975 / Beamline: ID23-1 / Wavelength: 0.91975 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Dec 4, 2004 / Details: TOROIDAL MIRROR |
| Radiation | Monochromator: SI (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91975 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→43.44 Å / Num. obs: 22693 / % possible obs: 99 % / Redundancy: 6.6 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 17 |
| Reflection shell | Resolution: 2.5→2.6 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.26 / Mean I/σ(I) obs: 5 / % possible all: 91 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1V05 Resolution: 2.5→43.44 Å / Cor.coef. Fo:Fc: 0.891 / Cor.coef. Fo:Fc free: 0.85 / SU B: 22.911 / SU ML: 0.248 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.552 / ESU R Free: 0.331 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. SOME OF THE SIDECHAIN ATOMS OF RESIDUES A 2051 ARG, A 2059 GLU, A 2074 ASP, A 2077 TYR, A 2089 LYS, A 2098 GLU, A 2123 GLN, A 2177 GLN, A ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. SOME OF THE SIDECHAIN ATOMS OF RESIDUES A 2051 ARG, A 2059 GLU, A 2074 ASP, A 2077 TYR, A 2089 LYS, A 2098 GLU, A 2123 GLN, A 2177 GLN, A 2187 GLU, A 2289 LYS B 2051 ARG, B 2058 HIS, B 2077 TYR, B 2098 GLU, B 2133 LYS, B2137 GLU, B 2149 ARG, B 2201 ARG, B 2202 PHE, B 2203 VAL, B 2206 GLU, B 2212 VAL, B 2228 PHE, B 2239 HIS, B 2240 LYS, B2280 LYS, B 2282 GLU, B 2289 LYS, B2290 ASP, B 2311 PHE AND B 2314 GLU HAVE A POORLY DEFINED ELECTRON DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.56 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→43.44 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|