 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2j0j: Crystal structure of a fragment of focal adhesion kinase containi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2j0j | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a fragment of focal adhesion kinase containing the FERM and kinase domains. | ||||||
 Components Components | FOCAL ADHESION KINASE 1 | ||||||
 Keywords Keywords | TRANSFERASE / CELL MIGRATION / FERM / INTEGRIN SIGNALING | ||||||
| Function / homology |  Function and homology information Function and homology informationApoptotic cleavage of cellular proteins / NCAM signaling for neurite out-growth / RAF/MAP kinase cascade / Turbulent (oscillatory, disturbed) flow shear stress activates signaling by PIEZO1 and integrins in endothelial cells / RHO GTPases Activate WASPs and WAVEs / Regulation of actin dynamics for phagocytic cup formation / negative regulation of protein autophosphorylation / radial glia-guided pyramidal neuron migration / positive regulation of protein tyrosine kinase activity / calcium-dependent cysteine-type endopeptidase activity ...Apoptotic cleavage of cellular proteins / NCAM signaling for neurite out-growth / RAF/MAP kinase cascade / Turbulent (oscillatory, disturbed) flow shear stress activates signaling by PIEZO1 and integrins in endothelial cells / RHO GTPases Activate WASPs and WAVEs / Regulation of actin dynamics for phagocytic cup formation / negative regulation of protein autophosphorylation / radial glia-guided pyramidal neuron migration / positive regulation of protein tyrosine kinase activity / calcium-dependent cysteine-type endopeptidase activity / positive regulation of substrate-dependent cell migration, cell attachment to substrate / Integrin signaling / GRB2:SOS provides linkage to MAPK signaling for Integrins / : / MET activates PTK2 signaling / Extra-nuclear estrogen signaling / EPHB-mediated forward signaling / p130Cas linkage to MAPK signaling for integrins / VEGFA-VEGFR2 Pathway / signal complex assembly / response to pH / angiogenesis involved in wound healing / wound healing, spreading of cells / positive regulation of protein binding / negative regulation of anoikis / negative regulation of cell-substrate adhesion / positive regulation of focal adhesion assembly / regulation of cell adhesion / response to muscle stretch / molecular function activator activity / actin filament organization / non-specific protein-tyrosine kinase / non-membrane spanning protein tyrosine kinase activity / sarcolemma / integrin binding / epidermal growth factor receptor signaling pathway / protein autophosphorylation / protein tyrosine kinase activity / protease binding / cell cortex / positive regulation of cell migration / ciliary basal body / focal adhesion / positive regulation of cell population proliferation / centrosome / perinuclear region of cytoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Lietha, D. / Cai, X. / Li, Y. / Schaller, M.D. / Eck, M.J. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2007 Title: Structural Basis for the Autoinhibition of Focal Adhesion Kinase Authors: Lietha, D. / Cai, X. / Ceccarelli, D.F.J. / Li, Y. / Schaller, M.D. / Eck, M.J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2j0j.cif.gz | 138.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2j0j.ent.gz | 106.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2j0j.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j0/2j0jftp://data.pdbj.org/pub/pdb/validation_reports/j0/2j0j | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2j0kC  2j0lC  2j0mC  1mp8S  2aehS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 75351.281 Da / Num. of mol.: 1 / Fragment: FERM AND KINASE DOMAINS, RESIDUES 31-686 Source method: isolated from a genetically manipulated source Details: UNPHOSPHORYLATED / Source: (gene. exp.)  TRICHOPLUSIA NI (cabbage looper) TRICHOPLUSIA NI (cabbage looper)References: UniProt: Q00944, non-specific protein-tyrosine kinase |
|---|---|
| #2: Chemical | ChemComp-4ST / 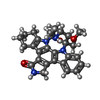  Mass: 470.563 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H30N4O3 Mass: 470.563 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H30N4O3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 22 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 22 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | RESIDUES 31-686 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.57 Å3/Da / Density % sol: 52.1 % |
|---|---|
| Crystal grow | pH: 8.5 Details: 13% PEG 10K, 250MM NACL, 100MM TRIS PH 8.5, 10MM TCEP. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 0.9795 / Beamline: 24-ID-C / Wavelength: 0.9795 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Aug 20, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→50 Å / Num. obs: 17892 / % possible obs: 91.2 % / Observed criterion σ(I): 0 / Redundancy: 5.7 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 18.3 |
| Reflection shell | Resolution: 2.8→2.9 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 2.84 / % possible all: 64.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1MP8 AND 2AEH Resolution: 2.8→45.55 Å / Cor.coef. Fo:Fc: 0.934 / Cor.coef. Fo:Fc free: 0.893 / SU B: 26.03 / SU ML: 0.348 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R Free: 0.437 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 68.63 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→45.55 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|