 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2hh5: Crystal Structure of Cathepsin S in complex with a Zinc mediated ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2hh5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Cathepsin S in complex with a Zinc mediated non-covalent arylaminoethyl amide | ||||||
 Components Components | Cathepsin S | ||||||
 Keywords Keywords | HYDROLASE / Cathepsin S / noncovalent / inhibition / zinc / arylaminoethyl-amides | ||||||
| Function / homology |  Function and homology information Function and homology informationcathepsin S / regulation of antigen processing and presentation / positive regulation of cation channel activity / basement membrane disassembly / antigen processing and presentation of peptide antigen / endolysosome lumen / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures ...cathepsin S / regulation of antigen processing and presentation / positive regulation of cation channel activity / basement membrane disassembly / antigen processing and presentation of peptide antigen / endolysosome lumen / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures / response to acidic pH / toll-like receptor signaling pathway / antigen processing and presentation / collagen catabolic process / fibronectin binding / extracellular matrix disassembly / Degradation of the extracellular matrix / collagen binding / phagocytic vesicle / cysteine-type peptidase activity / laminin binding / MHC class II antigen presentation / lysosomal lumen / : / Endosomal/Vacuolar pathway / protein processing / Degradation of CDH1 / antigen processing and presentation of exogenous peptide antigen via MHC class II / tertiary granule lumen / late endosome / peptidase activity / extracellular matrix / ficolin-1-rich granule lumen / adaptive immune response / lysosome / immune response / serine-type endopeptidase activity / cysteine-type endopeptidase activity / Neutrophil degranulation / proteolysis / : / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Spraggon, G. / Hornsby, M. / Lesley, S.A. / Tully, D.C. / Harris, J.L. / Karenewsky, D.S. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2006 Title: Synthesis and SAR of arylaminoethyl amides as noncovalent inhibitors of cathepsin S: P3 cyclic ethers. Authors: Tully, D.C. / Liu, H. / Chatterjee, A.K. / Alper, P.B. / Epple, R. / Williams, J.A. / Roberts, M.J. / Woodmansee, D.H. / Masick, B.T. / Tumanut, C. / Li, J. / Spraggon, G. / Hornsby, M. / ...Authors: Tully, D.C. / Liu, H. / Chatterjee, A.K. / Alper, P.B. / Epple, R. / Williams, J.A. / Roberts, M.J. / Woodmansee, D.H. / Masick, B.T. / Tumanut, C. / Li, J. / Spraggon, G. / Hornsby, M. / Chang, J. / Tuntland, T. / Hollenbeck, T. / Gordon, P. / Harris, J.L. / Karanewsky, D.S. #1: Journal: Bioorg.Med.Chem.Lett. / Year: 2006Title: Synthesis and evaluation of arylaminoethyl amides as noncovalent inhibitors of cathepsin S. Part 3: heterocyclic P3. Authors: Tully, D.C. / Liu, H. / Alper, P.B. / Chatterjee, A.K. / Epple, R. / Roberts, M.J. / Williams, J.A. / Nguyen, K.T. / Woodmansee, D.H. / Tumanut, C. / Li, J. / Spraggon, G. / Chang, J. / ...Authors: Tully, D.C. / Liu, H. / Alper, P.B. / Chatterjee, A.K. / Epple, R. / Roberts, M.J. / Williams, J.A. / Nguyen, K.T. / Woodmansee, D.H. / Tumanut, C. / Li, J. / Spraggon, G. / Chang, J. / Tuntland, T. / Harris, J.L. / Karanewsky, D.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2hh5.cif.gz | 107.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2hh5.ent.gz | 81.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2hh5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hh/2hh5ftp://data.pdbj.org/pub/pdb/validation_reports/hh/2hh5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2hhnC  2f1gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
| ||||||||||||
| Details | The biological assembly is a monomer - there are two monomers in the asymmetric unit. |
-Components
| #1: Protein | Mass: 24391.393 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSS / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P25774, cathepsin S Spodoptera frugiperda (fall armyworm) / References: UniProt: P25774, cathepsin S#2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn#3: Chemical |   Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Chemical | 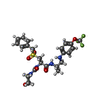  Mass: 572.597 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H31F3N4O6S Mass: 572.597 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H31F3N4O6S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 289 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 289 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.33 Å3/Da / Density % sol: 47.24 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 0.2M ZnAcetate, 20% PEG-3350, 150mM Sodium Chloride, pH 7.0, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.3 / Wavelength: 1 Å / Beamline: 5.0.3 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 28, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→34.2 Å / Num. all: 38549 / Num. obs: 38549 / % possible obs: 99.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.7 % / Rmerge(I) obs: 0.064 / Rsym value: 0.064 / Net I/σ(I): 17.7 |
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.763 / Mean I/σ(I) obs: 1.3 / Num. unique all: 4514 / Rsym value: 0.763 / % possible all: 98.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2F1G Resolution: 1.8→34.2 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.951 / SU B: 3.355 / SU ML: 0.101 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.127 / ESU R Free: 0.126 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.905 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→34.2 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.847 Å / Total num. of bins used: 20
|
