 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2da8: SOLUTION STRUCTURE OF A COMPLEX BETWEEN (N-MECYS3,N-MECYS7)TANDEM... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2da8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SOLUTION STRUCTURE OF A COMPLEX BETWEEN (N-MECYS3,N-MECYS7)TANDEM AND (D(GATATC))2 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA/ANTIBIOTIC / BISINTERCALATOR / DEPSIPEPTIDE / QUINOXALINE / ANTIBIOTIC / ANTITUMOR / DNA-ANTIBIOTIC COMPLEX | ||||||
| Function / homology | 2-CARBOXYQUINOXALINE / : / DNA Function and homology information Function and homology information | ||||||
| Biological species |  STREPTOMYCINEAE (bacteria) STREPTOMYCINEAE (bacteria) | ||||||
| Method | SOLUTION NMR / ENERGY MINIMIZATION, RESTRAINED MOLECULAR DYNAMICS, NOE BASED RELAXATION REFINEMENT | ||||||
 Authors Authors | Addess, K.J. / Sinsheimer, J.S. / Feigon, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1993 Title: Solution Structure of a Complex between [N-Mecys3,N-Mecys7]Tandem and [D(Gatatc)]2. Authors: Addess, K.J. / Sinsheimer, J.S. / Feigon, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2da8.cif.gz | 88.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2da8.ent.gz | 69.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2da8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/da/2da8ftp://data.pdbj.org/pub/pdb/validation_reports/da/2da8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 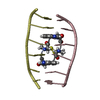
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Atom site foot note | 1: RESIDUES SER A 1 AND SER B 1 ARE D-AMINO ACIDS. 2: ALA A 2 - CYS A 3 MODEL 1 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: ALA B 2 - CYS B 3 MODEL 1 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: ALA A 2 - CYS A 3 MODEL 2 OMEGA = 0.02 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 5: ALA A 2 - CYS A 3 MODEL 3 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 6: ALA B 2 - CYS B 3 MODEL 3 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 7: ALA A 2 - CYS A 3 MODEL 4 OMEGA =359.98 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 8: ALA B 2 - CYS B 3 MODEL 4 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 9: ALA A 2 - CYS A 3 MODEL 5 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 10: ALA B 2 - CYS B 3 MODEL 5 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 11: ALA A 2 - CYS A 3 MODEL 6 OMEGA = 0.02 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 12: ALA A 2 - CYS A 3 MODEL 7 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 13: ALA A 2 - CYS A 3 MODEL 8 OMEGA = 0.00 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION | |||||||||
| NMR ensembles |
|
-Components
| #1: Protein/peptide | Mass: 766.928 Da / Num. of mol.: 1 / Mutation: YES / Source method: obtained synthetically / Details: VALINE AT POSITIONS 4 AND 8 / Source: (synth.) STREPTOMYCINEAE (bacteria) / References: NOR: NOR01129 | ||
|---|---|---|---|
| #2: DNA chain | Mass: 1808.229 Da / Num. of mol.: 2 / Source method: obtained synthetically #3: Chemical | 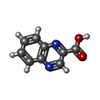  Mass: 174.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H6N2O2 Mass: 174.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H6N2O2 |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR details | Text: FOR DISTANCE GEOMETRY, FULL STRUCTURE EMBEDS WERE DONE USING THE PROGRAM DSPACE (HARE RESEARCH, INC.) AND SUBSTRUCTURE EMBEDS WERE DONE USING X-PLOR (A. T. BRUNGER, YALE UNIVERSITY). ALL ...Text: FOR DISTANCE GEOMETRY, FULL STRUCTURE EMBEDS WERE DONE USING THE PROGRAM DSPACE (HARE RESEARCH, INC.) AND SUBSTRUCTURE EMBEDS WERE DONE USING X-PLOR (A. T. BRUNGER, YALE UNIVERSITY). ALL REFINEMENT STEPS WERE DONE USING X-PLOR. |
- Processing
Processing
| Software |
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NMR software |
| |||||||||
| Refinement | Method: ENERGY MINIMIZATION, RESTRAINED MOLECULAR DYNAMICS, NOE BASED RELAXATION REFINEMENT Software ordinal: 1 Details: EIGHT STRUCTURES WERE GENERATED BY DISTANCE GEOMETRY. TWO DIMENSIONAL DERIVED NOE AND DIHEDRAL BOND ANGLE CONSTRAINTS WERE USED THROUGHOUT ALL STAGES OF THE STRUCTURE CALCULATIONS. AVERAGE R ...Details: EIGHT STRUCTURES WERE GENERATED BY DISTANCE GEOMETRY. TWO DIMENSIONAL DERIVED NOE AND DIHEDRAL BOND ANGLE CONSTRAINTS WERE USED THROUGHOUT ALL STAGES OF THE STRUCTURE CALCULATIONS. AVERAGE R VALUE FOR 8 STRUCTURES 0.21 | |||||||||
| NMR ensemble | Conformer selection criteria: all calculated structures submitted Conformers calculated total number: 8 / Conformers submitted total number: 8 | |||||||||
| NMR ensemble rms | Atom type: all heavy atoms / Distance rms dev: 1.1 Å |
 X-PLOR
X-PLOR