 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2b92: Crystal-structure of the N-terminal Large GTPase Domain of human ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2b92 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal-structure of the N-terminal Large GTPase Domain of human Guanylate Binding protein 1 (hGBP1) in complex with GDP/AlF3 | ||||||
 Components Components | Interferon-induced guanylate-binding protein 1 | ||||||
 Keywords Keywords | SIGNALING PROTEIN / protein- guanine nucleotide complex | ||||||
| Function / homology |  Function and homology information Function and homology informationRegulation of GBP-mediated host defense / GBP-mediated host defense / GDP phosphatase activity / non-canonical inflammasome complex assembly / negative regulation of substrate adhesion-dependent cell spreading / CASP4 inflammasome assembly / protein localization to vacuole / cytolysis in another organism / positive regulation of pyroptotic inflammatory response / symbiont cell surface ...Regulation of GBP-mediated host defense / GBP-mediated host defense / GDP phosphatase activity / non-canonical inflammasome complex assembly / negative regulation of substrate adhesion-dependent cell spreading / CASP4 inflammasome assembly / protein localization to vacuole / cytolysis in another organism / positive regulation of pyroptotic inflammatory response / symbiont cell surface / NS1 Mediated Effects on Host Pathways / vesicle membrane / Enterobacterial factors antagonize host defense / negative regulation of interleukin-2 production / defense response to protozoan / spectrin binding / cytokine binding / cellular response to interleukin-1 / negative regulation of T cell receptor signaling pathway / negative regulation of protein localization to plasma membrane / regulation of protein localization to plasma membrane / Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides / regulation of calcium-mediated signaling / cytoplasmic vesicle membrane / lipopolysaccharide binding / negative regulation of ERK1 and ERK2 cascade / Hsp90 protein binding / cellular response to type II interferon / cellular response to tumor necrosis factor / Interferon gamma signaling / GDP binding / actin cytoskeleton / actin binding / G protein activity / cytoplasmic vesicle / defense response to virus / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / defense response to bacterium / Golgi membrane / innate immune response / GTPase activity / GTP binding / Golgi apparatus / enzyme binding / protein homodimerization activity / extracellular region / identical protein binding / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å | ||||||
 Authors Authors | Ghosh, A. / Praefcke, G.J.K. / Renault, L. / Wittinghofer, A. / Herrmann, C. | ||||||
 Citation Citation | Journal: Nature / Year: 2006 Title: How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Authors: Ghosh, A. / Praefcke, G.J. / Renault, L. / Wittinghofer, A. / Herrmann, C. #1: Journal: Embo J. / Year: 2000Title: Triphosphate structure of guanylate-binding protein 1 and implications for nucleotide binding and GTPase mechanism. Authors: Prakash, B. / Renault, L. / Praefcke, G.J. / Herrmann, C. / Wittinghofer, A. #2: Journal: Nature / Year: 2000Title: Structure of human guanylate-binding protein 1 representing a unique class of GTP-binding proteins. Authors: Prakash, B. / Praefcke, G.J. / Renault, L. / Herrmann, C. / Wittinghofer, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2b92.cif.gz | 132.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2b92.ent.gz | 102.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2b92.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b9/2b92ftp://data.pdbj.org/pub/pdb/validation_reports/b9/2b92 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2b8wSC  2bc9C  2d4hC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | The biological assembly is the homodimer in the asymmetric unit |
-Components
| #1: Protein | Mass: 37078.449 Da / Num. of mol.: 2 / Fragment: N-terminal Large GTPase domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GBP1 / Plasmid: pQE9 / Species (production host): Escherichia coli / Production host:  #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical |   Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM#4: Chemical | 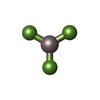  Mass: 83.977 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF3 Mass: 83.977 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.78 % |
|---|---|
| Crystal grow | Temperature: 298 K Details: 12.5% Peg3350, 125mM Na2HPO4 (from Hampton Research peg/ion screen supplied at pH9.1), VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.9393 / Beamline: ID14-4 / Wavelength: 0.9393 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 15, 2002 / Details: GE(220) CRYSTAL |
| Radiation | Monochromator: DIAMOND MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9393 Å / Relative weight: 1 |
| Reflection | Resolution: 3.2→30 Å / Num. obs: 14438 / % possible obs: 97.4 % / Observed criterion σ(I): -3 / Redundancy: 9.8 % / Biso Wilson estimate: 90.1 Å2 / Rsym value: 0.092 / Net I/σ(I): 17.9 |
| Reflection shell | Resolution: 3.2→3.26 Å / Redundancy: 11.7 % / Mean I/σ(I) obs: 6.8 / Rsym value: 0.457 / % possible all: 95.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2B8W Resolution: 3.2→30 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.925 / SU B: 41.573 / SU ML: 0.323 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.478 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 60.57 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.2→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.2→3.26 Å / Total num. of bins used: 27
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
