 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2a5p: Monomeric parallel-stranded DNA tetraplex with snap-back 3+1 3' G... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2a5p | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Monomeric parallel-stranded DNA tetraplex with snap-back 3+1 3' G-tetrad, single-residue chain reversal loops, GAG triad in the context of GAAG diagonal loop, NMR, 8 struct. | ||||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / MONOMERIC PARALLEL-STRANDED QUADRUPLEX / C-MYC PROMOTER 3+1 G-TETRAD / SINGLE NUCLEOTIDE CHAIN REVERSAL LOOP / GAG TRIAD / GAAG LOOP | Function / homology | DNA / DNA (> 10) |  Function and homology information Function and homology informationBiological species |  Homo sapiens (human) Homo sapiens (human)Method | SOLUTION NMR / DISTANCE GEOMETRY, SIMULATED ANNEALING, DISTANCE RESTRAINED MOLECULAR DYNAMICS REFINEMENT, RELAXATION MATRIX INTENSITY REFINEMENT |  Authors AuthorsPhan, A.T. / Kuryavyi, V.V. / Gaw, H.Y. / Patel, D.J. |  CitationJournal: Nat.Chem.Biol. / Year: 2005 CitationJournal: Nat.Chem.Biol. / Year: 2005Title: Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Authors: Phan, A.T. / Kuryavyi, V. / Gaw, H.Y. / Patel, D.J. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2a5p.cif.gz | 126.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2a5p.ent.gz | 103.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2a5p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a5/2a5pftp://data.pdbj.org/pub/pdb/validation_reports/a5/2a5p | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 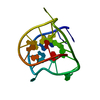
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: DNA chain | Mass: 7701.934 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: c-MYC gene / Source: (natural) Homo sapiens (human) |
|---|
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR experiment |
| ||||||||||||||||||||||||||||||||||||||||||||
| NMR details | Text: A TOTAL OF 100 INITIAL DNA STRUCTURES WERE GENERATED USING THE METRIC MATRIX DISTANCE GEOMETRY PROTOCOL WITH THE EXPERIMENTAL DISTANCE RESTRAINTS SPECIFIED WITH THE R-6 AVERAGING OPTION. EIGHT ...Text: A TOTAL OF 100 INITIAL DNA STRUCTURES WERE GENERATED USING THE METRIC MATRIX DISTANCE GEOMETRY PROTOCOL WITH THE EXPERIMENTAL DISTANCE RESTRAINTS SPECIFIED WITH THE R-6 AVERAGING OPTION. EIGHT BEST STRUCTURES SELECTED ON THE BASIS OF GOOD COVALENT GEOMETRY, LOW DISTANCE RESTRAINT VIOLATION AND FAVOURABLE NON-BONDED ENERGY TERMS WERE FURTHER OPTIMIZED WITH RESTRAINED MOLECULAR DYNAMICS, MOLECULAR MECHANICS AND RELAXATION MATRIX INTENSITY REFINEMENT.THE PROTOCOLS ARE AS FOLLOWS. THE DYNAMICS WAS INITIATED AT 300K AND THE TEMPERATURE WAS INCREASED TO 1000K DURING 7PS. AFTER 0.5 PS THE FORCE CONSTANTS WERE GRADUALLY SCALED DURING 17.5 PSEC IN THE PROPORTION 1:4:8 FOR THREE CLASSES OF NOE: EXCHANGEABLE, NONEXCHANGEABLE AND HYDROGEN BONDS, RESP. THE STRUCTURES WERE THEN SLOWLY COOLED TO 300K IN 14 PS AND EQUILIBRATED AT 300K FOR 12 PS. THE COORDINATES SAVED EVERY 0.5 PS DURING THE LAST 4.0 PS WERE AVERAGED AND THE AVERAGE STRUCTURE WAS SUBJECTED TO MINIMIZATION UNTIL THE GRADIENT OF ENERGY WAS LESS THAN 0.1KCAL/MOL. SOFT PLANARITY RESTRAINTS IMPOSED ON TETRADS AND TRIAD BEFORE HEATING WERE EXCLUDED AT THE EQUILIBRATION STAGE. THE DIHEDRAL AND HYDROGEN-BONDING RESTRAINTS FOR TETRADS, WERE MAINTAINED THROUGHOUT. ALL EIGHT STRUCTURES WERE SUBJECTED TO RELAXATION MATRIX INTENSITY REFINEMENT. THE INTENSITY VOLUMES OF NON-EXCHANGEABLE PROTONS WERE USED AS RESTRAINTS WITH EXCHANGEABLE PROTONS REPLACED BY DEUTERONS. DYNAMICS STARTED AT 5K AND THE SYSTEM WAS HEATHED UP TO 300 K IN 0.6 PSEC. THE FORCE CONSTANT FOR NOE INTENSITIES WAS GRADUALLY INCREASED FROM 0 TO 400 KCAL*MOL-1*A-2 WITH SIMULTANEOUS DECREASE OF THE DISTANCE FORCE CONSTANTS FOR NON-EXCHANGEABLE PROTONS FROM 32 to 16. AFTER EQUILIBRATION AT 300 K FOR 3 PSEC THE RESULTING STRUCTURES WERE SUBJECTED TO MINIMIZATION UNTIL THE GRADIENT OF ENERGY WAS LESS THAN 0.1KCAL/MOL. THE DISTANCE RESTRAINTS FOR EXCHANGEABLE PROTONS HYDROGEN BONDS AND DIHEDRAL ANGLE RESTRAINTS WERE MAINTAINED. SOFT PLANARITY RESTRAINTS WEERE SET FOR TETRAD (1 KCAL*MOL-1*A-2) AND TRIAD (0.5 KCAL*MOL-1*A-2). |
 HSQC
HSQC- Sample preparation
Sample preparation
| Details |
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample conditions | Ionic strength: 90 mM / pH: 7 / Pressure: 1 atm / Temperature: 298 K |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radiation wavelength | Relative weight: 1 | |||||||||||||||
| NMR spectrometer |
|
- Processing
Processing
| NMR software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: DISTANCE GEOMETRY, SIMULATED ANNEALING, DISTANCE RESTRAINED MOLECULAR DYNAMICS REFINEMENT, RELAXATION MATRIX INTENSITY REFINEMENT Software ordinal: 1 | ||||||||||||||||
| NMR representative | Selection criteria: lowest energy | ||||||||||||||||
| NMR ensemble | Conformer selection criteria: back calculated data agree with experimental NOESY spectrum Conformers calculated total number: 100 / Conformers submitted total number: 8 |
