DNA synthesis involved in DNA replication / DNA exonuclease activity / viral DNA genome replication / Hydrolases; Acting on ester bonds; Exodeoxyribonucleases producing 5'-phosphomonoesters / DNA polymerase processivity factor activity / protein-disulfide reductase activity / 3'-5' exonuclease activity / cell redox homeostasis / DNA-templated DNA replication / double-strand break repair ...DNA synthesis involved in DNA replication / DNA exonuclease activity / viral DNA genome replication / Hydrolases; Acting on ester bonds; Exodeoxyribonucleases producing 5'-phosphomonoesters / DNA polymerase processivity factor activity / protein-disulfide reductase activity / 3'-5' exonuclease activity / cell redox homeostasis / DNA-templated DNA replication / double-strand break repair / DNA-directed DNA polymerase / DNA-directed DNA polymerase activity / nucleotide binding / DNA binding / metal ion binding / cytosol / cytoplasm Similarity search - Function
DNA-directed DNA polymerase T7 / Taq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / Thioredoxin / DNA polymerase A / DNA polymerase family A / DNA-directed DNA polymerase, family A, conserved site / DNA polymerase family A signature. / DNA-directed DNA polymerase, family A, palm domain ...DNA-directed DNA polymerase T7 / Taq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / Thioredoxin / DNA polymerase A / DNA polymerase family A / DNA-directed DNA polymerase, family A, conserved site / DNA polymerase family A signature. / DNA-directed DNA polymerase, family A, palm domain / DNA polymerase A domain / Thioredoxin / Thioredoxin family active site. / Thioredoxin, conserved site / Thioredoxin domain profile. / Thioredoxin domain / 5' to 3' exonuclease, C-terminal subdomain / Ribonuclease H-like superfamily/Ribonuclease H / Glutaredoxin / Glutaredoxin / DNA polymerase; domain 1 / Nucleotidyltransferase; domain 5 / Thioredoxin-like superfamily / Ribonuclease H superfamily / Ribonuclease H-like superfamily / Alpha-Beta Plaits / DNA/RNA polymerase superfamily / Up-down Bundle / 2-Layer Sandwich / Orthogonal Bundle / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
2',3'-DIDEOXYADENOSINE-5'-TRIPHOSPHATE / DNA / DNA (> 10) / DNA-directed DNA polymerase / Thioredoxin 1 Similarity search - Component
Biological species
Enterobacteria phage T7 (virus) Escherichia coli (E. coli)
Mass: 18.015 Da / Num. of mol.: 219 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.878 Å3/Da / Density % sol: 57.2 %
Crystal grow
Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG, amonium sulfate, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K
-
Data collection
Diffraction
ID
Mean temperature (K)
Crystal-ID
1
200
1
2
200
1
Diffraction source
Source
Site
Beamline
ID
Wavelength (Å)
SYNCHROTRON
NSLS
X26C
1
1
ROTATING ANODE
2
1.4
Detector
Type
ID
Detector
Date
ADSC QUANTUM 4
1
CCD
Mar 30, 2005
2
IMAGE PLATE
Jan 15, 2005
Radiation
ID
Protocol
Monochromatic (M) / Laue (L)
Scattering type
Wavelength-ID
1
SINGLEWAVELENGTH
M
x-ray
1
2
SINGLEWAVELENGTH
M
x-ray
2
Radiation wavelength
ID
Wavelength (Å)
Relative weight
1
1
1
2
1.4
1
Reflection
Resolution: 2.5→50 Å / Num. obs: 39043 / Biso Wilson estimate: 38.4 Å2
Reflection shell
Resolution: 2.5→2.59 Å / Num. unique all: 39043
-
Processing
Software
Name
Version
Classification
CNS
1.1
refinement
HKL-2000
datareduction
SCALEPACK
datascaling
CNS
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.7→47.6 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1895166.73 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: The authors state that the unusual peptide bond lengths for K293-V294 (in thioredoxin binding loop) and V541-G542 (in the fingers subdomain) may reflect inherent problems with model ...Details: The authors state that the unusual peptide bond lengths for K293-V294 (in thioredoxin binding loop) and V541-G542 (in the fingers subdomain) may reflect inherent problems with model refinement resulting from local disorder in these regions of the structure, which are known to be mobile in different structures of T7 DNA polymerase.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Enterobacteria phage T7 (virus)
Enterobacteria phage T7 (virus)
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj







































 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
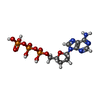
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 475.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O11P3
Mass: 475.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O11P3 Sample preparation
Sample preparation
 Processing
Processing