 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1skn: THE BINDING DOMAIN OF SKN-1 IN COMPLEX WITH DNA: A NEW DNA-BINDIN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1skn | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE BINDING DOMAIN OF SKN-1 IN COMPLEX WITH DNA: A NEW DNA-BINDING MOTIF | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / COMPLEX (TRANSCRIPTION FACTOR-DNA) / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of cellular response to oxidative stress / positive regulation of cellular response to manganese ion / Nuclear events mediated by NFE2L2 / GSK3B and BTRC:CUL1-mediated-degradation of NFE2L2 / Neddylation / KEAP1-NFE2L2 pathway / ATF6-mediated unfolded protein response / mesendoderm development / endodermal cell fate specification / response to paraquat ...positive regulation of cellular response to oxidative stress / positive regulation of cellular response to manganese ion / Nuclear events mediated by NFE2L2 / GSK3B and BTRC:CUL1-mediated-degradation of NFE2L2 / Neddylation / KEAP1-NFE2L2 pathway / ATF6-mediated unfolded protein response / mesendoderm development / endodermal cell fate specification / response to paraquat / embryonic digestive tract development / embryonic pattern specification / response to superoxide / cell fate specification / digestive tract development / cellular detoxification / IRE1-mediated unfolded protein response / Hsp70 protein binding / determination of adult lifespan / RNA polymerase II transcription regulatory region sequence-specific DNA binding / regulation of translation / response to heat / regulation of gene expression / response to oxidative stress / sequence-specific DNA binding / defense response to Gram-negative bacterium / DNA-binding transcription factor activity, RNA polymerase II-specific / defense response to bacterium / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / positive regulation of gene expression / regulation of transcription by RNA polymerase II / endoplasmic reticulum / DNA-templated transcription / positive regulation of transcription by RNA polymerase II / mitochondrion / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 2.5 Å | ||||||
 Authors Authors | Rupert, P.B. / Daughdrill, G.W. / Bowerman, B. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1998 Title: A new DNA-binding motif in the Skn-1 binding domain-DNA complex. Authors: Rupert, P.B. / Daughdrill, G.W. / Bowerman, B. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1skn.cif.gz | 48 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1skn.ent.gz | 30.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1skn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sk/1sknftp://data.pdbj.org/pub/pdb/validation_reports/sk/1skn | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 4528.961 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 4649.033 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #3: Protein | Mass: 10996.467 Da / Num. of mol.: 1 / Fragment: BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
| #4: Chemical | ChemComp-LDA / 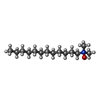  Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 28 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 28 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6 Details: PROTEIN WAS CRYSTALLIZED FROM 8% PEG 1000 50 MM BIS-TRIS, PH 6.0, 100-150 MM LICL, 2MM LDAO., VAPOR DIFFUSION, HANGING DROP, temperature 277.00K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Beamline: BL7-1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 15, 1997 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.5→20 Å / Num. obs: 7202 / % possible obs: 94.8 % / Redundancy: 6.9 % / Biso Wilson estimate: 38.4 Å2 / Rmerge(I) obs: 0.038 / Rsym value: 0.038 |
| Reflection shell | Resolution: 2.5→2.57 Å / Redundancy: 6.9 % / Rmerge(I) obs: 0.116 / % possible all: 100 |
| Reflection | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 20 Å |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIRAS / Resolution: 2.5→20 Å / Cross valid method: THROUGHOUT Details: THE SHORT CONTACTS LISTED IN REMARK 500 ARE THE RESULT OF THE LAURYLDIMETHYLAMINEOXIDE (LDA) MOLECULE MODELED ON A TWO-FOLD AXIS AT HALF OCCUPANCY. THIS LDA MOLECULE WAS EXCLUDED FROM MAKING ...Details: THE SHORT CONTACTS LISTED IN REMARK 500 ARE THE RESULT OF THE LAURYLDIMETHYLAMINEOXIDE (LDA) MOLECULE MODELED ON A TWO-FOLD AXIS AT HALF OCCUPANCY. THIS LDA MOLECULE WAS EXCLUDED FROM MAKING BAD CONTACTS WITH A SYMMETRY-RELATED COPY OF ITSELF IN THE TNT REFINEMENT PROTOCOL.
| ||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: TNT 5F / Bsol: 110 Å2 / ksol: 0.8 e/Å3 | ||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→20 Å
| ||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Version: 5F / Classification: refinement | ||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.225 | ||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
