 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1rqq: Crystal Structure of the Insulin Receptor Kinase in Complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1rqq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Insulin Receptor Kinase in Complex with the SH2 Domain of APS | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/SIGNALING PROTEIN / Protein tyrosine kinase / adaptor protein / SH2 domain / TRANSFERASE-SIGNALING PROTEIN COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationRegulation of KIT signaling / Factors involved in megakaryocyte development and platelet production / antigen receptor-mediated signaling pathway / regulation of Ras protein signal transduction / B-1 B cell homeostasis / regulation of female gonad development / positive regulation of meiotic cell cycle / insulin-like growth factor II binding / positive regulation of developmental growth / male sex determination ...Regulation of KIT signaling / Factors involved in megakaryocyte development and platelet production / antigen receptor-mediated signaling pathway / regulation of Ras protein signal transduction / B-1 B cell homeostasis / regulation of female gonad development / positive regulation of meiotic cell cycle / insulin-like growth factor II binding / positive regulation of developmental growth / male sex determination / insulin receptor complex / insulin-like growth factor I binding / insulin receptor activity / transmembrane receptor protein tyrosine kinase adaptor activity / positive regulation of protein-containing complex disassembly / exocrine pancreas development / dendritic spine maintenance / insulin binding / adrenal gland development / cargo receptor activity / PTB domain binding / Signaling by Insulin receptor / IRS activation / neuronal cell body membrane / positive regulation of respiratory burst / amyloid-beta clearance / regulation of embryonic development / insulin receptor substrate binding / positive regulation of receptor internalization / epidermis development / positive regulation of glycogen biosynthetic process / Signal attenuation / regulation of immune response / protein kinase activator activity / transport across blood-brain barrier / heart morphogenesis / phosphatidylinositol 3-kinase binding / Insulin receptor recycling / insulin-like growth factor receptor binding / ruffle / stress fiber / neuron projection maintenance / positive regulation of mitotic nuclear division / signaling adaptor activity / Insulin receptor signalling cascade / SH2 domain binding / receptor-mediated endocytosis / dendrite membrane / brown fat cell differentiation / positive regulation of glycolytic process / positive regulation of D-glucose import across plasma membrane / B cell receptor signaling pathway / learning / actin filament / receptor protein-tyrosine kinase / caveola / receptor internalization / male gonad development / cellular response to growth factor stimulus / cellular response to insulin stimulus / cytokine-mediated signaling pathway / memory / positive regulation of nitric oxide biosynthetic process / insulin receptor signaling pathway / protein autophosphorylation / late endosome / nervous system development / glucose homeostasis / amyloid-beta binding / PI5P, PP2A and IER3 Regulate PI3K/AKT Signaling / actin cytoskeleton organization / protein tyrosine kinase activity / positive regulation of canonical NF-kappaB signal transduction / positive regulation of MAPK cascade / lysosome / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / signaling receptor complex / endosome membrane / intracellular signal transduction / positive regulation of cell migration / G protein-coupled receptor signaling pathway / external side of plasma membrane / protein domain specific binding / axon / positive regulation of cell population proliferation / symbiont entry into host cell / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / GTP binding / protein-containing complex binding / extracellular exosome / ATP binding / membrane / identical protein binding / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Hu, J. / Liu, J. / Ghirlando, R. / Saltiel, A.R. / Hubbard, S.R. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 2003 Title: Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Authors: Hu, J. / Liu, J. / Ghirlando, R. / Saltiel, A.R. / Hubbard, S.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1rqq.cif.gz | 171.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1rqq.ent.gz | 133.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1rqq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rq/1rqqftp://data.pdbj.org/pub/pdb/validation_reports/rq/1rqq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rpySC  1gagS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 4 molecules ABCD
| #1: Protein | Mass: 35033.660 Da / Num. of mol.: 2 / Fragment: Kinase domain / Mutation: C980S, K1251N Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: INSR / Cell line (production host): SF9 / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P06213, EC: 2.7.1.112 Spodoptera frugiperda (fall armyworm) / References: UniProt: P06213, EC: 2.7.1.112#2: Protein | Mass: 12891.678 Da / Num. of mol.: 2 / Fragment: SH2 domain Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
-Protein/peptide , 1 types, 2 molecules EF
| #3: Protein/peptide | Mass: 1939.280 Da / Num. of mol.: 2 / Source method: obtained synthetically |
|---|
-Non-polymers , 3 types, 83 molecules 
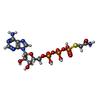

| #4: Chemical |  Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#5: Chemical |  Mass: 580.298 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N6O13P3S Mass: 580.298 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N6O13P3S#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 79 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.57 Å3/Da / Density % sol: 56 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: pH 8.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 / Beamline: X25 / Wavelength: 1.1 |
| Detector | Detector: CCD / Date: Nov 3, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→50 Å / Num. obs: 32437 / % possible obs: 99.8 % / Observed criterion σ(I): 0 / Redundancy: 4.8 % / Rsym value: 0.039 / Net I/σ(I): 22.4 |
| Reflection shell | Resolution: 2.6→2.69 Å / Rsym value: 0.164 / % possible all: 99.4 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. measured all: 156301 / Rmerge(I) obs: 0.039 |
| Reflection shell | *PLUS % possible obs: 99.4 % / Rmerge(I) obs: 0.164 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1GAG, 1RPY Resolution: 2.6→30 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS / Bsol: 31.06 Å2 / ksol: 0.32 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 49.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 30 Å / Num. reflection obs: 31461 / % reflection Rfree: 5 % / Rfactor Rfree: 0.279 / Rfactor Rwork: 0.233 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
