 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pxv: The staphostatin-staphopain complex: a forward binding inhibitor ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pxv | ||||||
|---|---|---|---|---|---|---|---|
| Title | The staphostatin-staphopain complex: a forward binding inhibitor in complex with its target cysteine protease | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / cysteine protease inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationcysteine-type endopeptidase inhibitor activity / cysteine-type peptidase activity / Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases / proteolysis / extracellular region / cytoplasm Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Filipek, R. / Rzychon, M. / Oleksy, A. / Gruca, M. / Dubin, A. / Potempa, J. / Bochtler, M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: The Staphostatin-Staphopain Complex: A FORWARD BINDING INHIBITOR IN COMPLEX WITH ITS TARGET CYSTEINE PROTEASE. Authors: Filipek, R. / Rzychon, M. / Oleksy, A. / Gruca, M. / Dubin, A. / Potempa, J. / Bochtler, M. #1: Journal: Protein Sci. / Year: 2003Title: Staphostatins resemble lipocalins, not cystatins in fold. Authors: Rzychon, M. / Filipek, R. / Sabat, A. / Kosowska, K. / Dubin, A. / Potempa, J. / Bochtler, M. #2: Journal: MOL.MICROBIOL. / Year: 2003Title: Staphostatins: an expanding new group of proteinase inhibitors with a unique specificity for the regulation of staphopains, Staphylococcus spp. cysteine proteinases Authors: Rzychon, M. / Sabat, A. / Kosowska, K. / Potempa, J. / Dubin, A. #3: Journal: J.Biol.Chem. / Year: 2002Title: Identification of a novel maturation mechanism and restricted substrate specificity for the SspB cysteine protease of Staphylococcus aureus Authors: Massimi, I. / Park, E. / Rice, K. / Muller-Esterl, W. / Sauder, D. / McGavin, M.J. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE At the time of processing, there was no database sequence available for the proteins from ...SEQUENCE At the time of processing, there was no database sequence available for the proteins from Staphylococcus aureus, strain V8 that were crystallized here. The closest homologues with protein sequences in a database were from Staphylococcus aureus subsp. aureus MW2. The author claims that the residue conflicts between strain V8 and strain MW2 noted here are genuine, confirmed strain differences. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pxv.cif.gz | 143 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pxv.ent.gz | 110.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1pxv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/px/1pxvftp://data.pdbj.org/pub/pdb/validation_reports/px/1pxv | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit |
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 |
| ||||||||
| 3 |
| ||||||||
| 4 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21062.078 Da / Num. of mol.: 2 / Mutation: C243A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: staphopain B / Production host: References: UniProt: Q70UQ8, UniProt: P0C1S6*PLUS, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases #2: Protein | Mass: 13074.622 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: staphostatin B / Production host: References: UniProt: Q9EYW6, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GAI / | 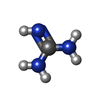  Mass: 59.070 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH5N3 Mass: 59.070 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH5N3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 458 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 458 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 56.61 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, sitting drop / pH: 6.3 Details: 2 M (NH4)2SO4 and 5% isopropanol, 100 mM guanidinium hydrochloride, pH 6.3, VAPOR DIFFUSION, SITTING DROP, temperature 294K | |||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.05 Å / Beamline: BW6 / Wavelength: 1.05 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jan 20, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.05 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→20.92 Å / Num. all: 72496 / Num. obs: 66436 / % possible obs: 94.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Biso Wilson estimate: 35 Å2 / Rmerge(I) obs: 0.052 / Rsym value: 0.052 / Net I/σ(I): 9.8 |
| Reflection shell | Resolution: 1.8→1.9 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.182 / Mean I/σ(I) obs: 3.5 / Num. unique all: 3959 / Rsym value: 0.182 / % possible all: 89.3 |
| Reflection | *PLUS Num. obs: 64796 / % possible obs: 92 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1CV8, 1NYC Resolution: 1.8→10 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.947 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.123 / ESU R Free: 0.117 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.263 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.845 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Rfactor Rfree: 0.221 / Rfactor Rwork: 0.192 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
