 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1rgz: Enterobacter cloacae GC1 Class C beta-Lactamase Complexed with Tr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1rgz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Enterobacter cloacae GC1 Class C beta-Lactamase Complexed with Transition-State Analog of Cefotaxime | ||||||
 Components Components | class C beta-lactamase | ||||||
 Keywords Keywords | HYDROLASE / cephalosporinase / enzyme inhibition / phosphonate / beta-lactam antibiotics / drug design | ||||||
| Function / homology |  Function and homology information Function and homology informationantibiotic catabolic process / beta-lactamase / beta-lactamase activity / outer membrane-bounded periplasmic space / response to antibiotic Similarity search - Function | ||||||
| Biological species |  Enterobacter cloacae (bacteria) Enterobacter cloacae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.37 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.37 Å | ||||||
 Authors Authors | Nukaga, M. / Kumar, S. / Nukaga, K. / Pratt, R.F. / Knox, J.R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2004 Title: Hydrolysis of third-generation cephalosporins by class C beta-lactamases. Structures of a transition state analog of cefotoxamine in wild-type and extended spectrum enzymes. Authors: Nukaga, M. / Kumar, S. / Nukaga, K. / Pratt, R.F. / Knox, J.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1rgz.cif.gz | 173 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1rgz.ent.gz | 136.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1rgz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rg/1rgzftp://data.pdbj.org/pub/pdb/validation_reports/rg/1rgz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rgyC  1gceS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 39508.062 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacter cloacae (bacteria) / Gene: BLA / Plasmid: pHSG398 / Production host: | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-PTX / {[(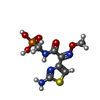  Mass: 294.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H11N4O5PS Mass: 294.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H11N4O5PS | ||||
| #3: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 398 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 398 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.32 % |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, sitting drop / pH: 7 Details: PEG 8000, HEPES, pH 7.0, VAPOR DIFFUSION, SITTING DROP, temperature 296K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.9474 Å / Beamline: A1 / Wavelength: 0.9474 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jun 24, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9474 Å / Relative weight: 1 |
| Reflection | Resolution: 1.37→50 Å / Num. all: 64695 / Num. obs: 64695 / % possible obs: 96 % / Observed criterion σ(I): 11.1 / Redundancy: 5 % / Rsym value: 0.096 / Net I/σ(I): 11.1 |
| Reflection shell | Resolution: 1.37→1.42 Å / Redundancy: 3.2 % / Mean I/σ(I) obs: 2.7 / Num. unique all: 5154 / Rsym value: 0.421 / % possible all: 77.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1GCE Resolution: 1.37→20 Å / Num. parameters: 29748 / Num. restraintsaints: 37290 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: The author notes that residues 219 to 225 in the omega loop, have 2 conformations (with 0.75 and 0.25 occupancies, conformers A and B respectively). Only the higher occupancy conformer (A) ...Details: The author notes that residues 219 to 225 in the omega loop, have 2 conformations (with 0.75 and 0.25 occupancies, conformers A and B respectively). Only the higher occupancy conformer (A) binds the phosphonate ligand (PTX). The preceeding residue (218) is also only linked to conformer A of residue 219. Likewise, there are two sets of water molecules in the binding site with corresponding occupancies. Therefore some waters appear to overlap with each other or with the ligand and are listed as close contacts in remark 500.
| |||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.8 Å2 | |||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.1 Å / Num. disordered residues: 34 / Occupancy sum hydrogen: 2675 / Occupancy sum non hydrogen: 3178.25 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.37→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
