 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qpb: PYRUVATE DECARBOYXLASE FROM YEAST (FORM B) COMPLEXED WITH PYRUVAMIDE -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qpb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | PYRUVATE DECARBOYXLASE FROM YEAST (FORM B) COMPLEXED WITH PYRUVAMIDE | |||||||||
 Components Components | PYRUVATE DECARBOXYLASE (FORM B) | |||||||||
 Keywords Keywords | LYASE / THIAMINE PYRUVATE / PYRUVAMIDE | |||||||||
| Function / homology |  Function and homology information Function and homology informationphenylpyruvate decarboxylase / branched-chain-2-oxoacid decarboxylase / phenylpyruvate decarboxylase activity / branched-chain-2-oxoacid decarboxylase activity / indolepyruvate decarboxylase / pyruvate fermentation to ethanol / indolepyruvate decarboxylase activity / : / pyruvate decarboxylase activity / Lyases; Carbon-carbon lyases; Carboxy-lyases ...phenylpyruvate decarboxylase / branched-chain-2-oxoacid decarboxylase / phenylpyruvate decarboxylase activity / branched-chain-2-oxoacid decarboxylase activity / indolepyruvate decarboxylase / pyruvate fermentation to ethanol / indolepyruvate decarboxylase activity / : / pyruvate decarboxylase activity / Lyases; Carbon-carbon lyases; Carboxy-lyases / L-tryptophan catabolic process / branched-chain amino acid catabolic process / L-phenylalanine catabolic process / thiamine pyrophosphate binding / magnesium ion binding / nucleus / cytoplasm / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | |||||||||
 Authors Authors | Lu, G. / Dobritzsch, D. / Schneider, G. | |||||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 2000 Title: The Structural Basis of Substrate Activation in Yeast Pyruvate Decarboxylase a Crystallographic and Kinetic Study Authors: Lu, G. / Dobritzsch, D. / Baumann, S. / Schneider, G. / Konig, S. #1: Journal: FEBS Lett. / Year: 1997 Title: Novel Tetramer Assembly of Pyruvate Decarboxylase from Brewer'S Yeast Observed in a New Crystal Form Authors: Lu, G. / Dobritzsch, D. / Konig, S. / Schneider, G. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX DETERMINATION METHOD: DSSP | |||||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qpb.cif.gz | 226.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qpb.ent.gz | 180.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1qpb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qp/1qpbftp://data.pdbj.org/pub/pdb/validation_reports/qp/1qpb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1pvdS S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 61555.074 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: FORM B / Source: (natural) #2: Chemical | 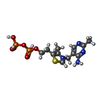  Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical | 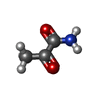  Mass: 87.077 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5NO2 Mass: 87.077 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5NO2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 141 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 141 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 43 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.8 Details: 8-14% PEG6000 20MM SODIUM CITRATE AT PH 5.7 1MM DTE, 5MM THDP, 5MM MGSO4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 2, 1997 |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→15 Å / Num. obs: 42140 / % possible obs: 88.9 % / Observed criterion σ(I): 1 / Redundancy: 4.3 % / Biso Wilson estimate: 33.0519 Å2 / Rmerge(I) obs: 0.108 / Rsym value: 0.091 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 2.4→2.46 Å / Redundancy: 3.5 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 5 / Rsym value: 0.38 / % possible all: 60 |
| Reflection shell | *PLUS % possible obs: 56 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PVD Resolution: 2.4→15 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 15 / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT CORRECTION WAS APPLIED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.0519 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.23 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINED | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.38→2.42 Å / Rfactor Rfree error: 0.04 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 3.8 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
