 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pyd: CATALYTIC CENTERS IN THE THIAMIN DIPHOSPHATE DEPENDENT ENZYME PYR... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pyd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CATALYTIC CENTERS IN THE THIAMIN DIPHOSPHATE DEPENDENT ENZYME PYRUVATE DECARBOXYLASE AT 2.4 ANGSTROMS RESOLUTION | |||||||||
 Components Components | PYRUVATE DECARBOXYLASE | |||||||||
 Keywords Keywords | LYASE(CARBON-CARBON) | |||||||||
| Function / homology |  Function and homology information Function and homology informationphenylpyruvate decarboxylase / branched-chain-2-oxoacid decarboxylase / phenylpyruvate decarboxylase activity / branched-chain-2-oxoacid decarboxylase activity / indolepyruvate decarboxylase / pyruvate fermentation to ethanol / indolepyruvate decarboxylase activity / : / pyruvate decarboxylase activity / Lyases; Carbon-carbon lyases; Carboxy-lyases ...phenylpyruvate decarboxylase / branched-chain-2-oxoacid decarboxylase / phenylpyruvate decarboxylase activity / branched-chain-2-oxoacid decarboxylase activity / indolepyruvate decarboxylase / pyruvate fermentation to ethanol / indolepyruvate decarboxylase activity / : / pyruvate decarboxylase activity / Lyases; Carbon-carbon lyases; Carboxy-lyases / branched-chain amino acid catabolic process / L-tryptophan catabolic process / L-phenylalanine catabolic process / thiamine pyrophosphate binding / magnesium ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.4 Å X-RAY DIFFRACTION / Resolution: 2.4 Å | |||||||||
 Authors Authors | Furey, W. / Dyda, F. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 1993 Title: Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4-A resolution. Authors: Dyda, F. / Furey, W. / Swaminathan, S. / Sax, M. / Farrenkopf, B. / Jordan, F. #1: Journal: Biochemistry and Physiology of Thiamin Diphosphate EnzymesYear: 1991 Title: Multiple Crystal Forms of Brewers' Yeast Pyruvate Decarboxylase: Characterization and Preliminary Crystallographic Analysis Authors: Dyda, F. / Furey, W. / Swaminathan, S. / Sax, M. / Jordan, F. / Farrenkopf, B. #2: Journal: J.Biol.Chem. / Year: 1990Title: Preliminary Crystallographic Data for the Thiamin Diphosphate-Dependent Enzyme Pyruvate Decarboxylase from Brewers' Yeast Authors: Dyda, F. / Furey, W. / Swaminathan, S. / Sax, M. / Farrenkopf, B. / Jordan, F. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pyd.cif.gz | 217.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pyd.ent.gz | 169.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1pyd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/py/1pydftp://data.pdbj.org/pub/pdb/validation_reports/py/1pyd | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.91229, -0.04516, -0.40704), Vector: Details | THE UNIT CELL CONTAINS TWO TETRAMERS, WITH ONE DIMER IN THE ASYMMETRIC UNIT. IN TERMS OF THE CRYSTAL AXES, THE TWO MONOMERS IN THE ASYMMETRIC UNIT (CHAINS *A* AND *B*) ARE RELATED BY A NONCRYSTALLOGRAPHIC TWO-FOLD AXIS INCLINED BY 83.31 DEGREES TO THE B AXIS, AND WHOSE PROJECTION ON THE AC PLANE MAKES ANGLES OF 102.16 DEGREES WITH THE A AXIS AND 14.23 DEGREES WITH THE C AXIS. THIS TRANSFORMATION IS PRESENTED ON *MTRIX* RECORDS BELOW. THE COMPLETE TETRAMER IS FORMED BY THE SUBMITTED DIMER AND ITS CRYSTALLOGRAPHICALLY RELATED MATE OBTAINED BY TWO-FOLD ROTATION ABOUT THE B AXIS. THE NONCRYSTALLOGRAPHIC TWO-FOLD AXIS DOES NOT INTERSECT THE B AXIS, AS ITS CLOSEST APPROACH IS OFFSET BY 1.352 ANGSTROMS. THE TETRAMER THUS HAS ONLY APPROXIMATE 222 SYMMETRY, AS IT MUST INTERSECT THE B AXIS AND BE INCLINED TO IT BY 90 DEGREES FOR EXACT 222 SYMMETRY. | |
-Components
| #1: Protein | Mass: 60872.344 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P06169, pyruvate decarboxylase #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 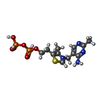  Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2OCompound details | THE CATALYTIC CENTERS ARE LOCATED AT THE INTERFACE BETWEEN MONOMERS WITHIN EACH DIMER, WITH TWO ...THE CATALYTIC CENTERS ARE LOCATED AT THE INTERFACE BETWEEN MONOMERS WITHIN EACH DIMER, WITH TWO SITES PER DIMER AND FOUR PER TETRAMER. | Sequence details | SINCE THE SEQUENCE FOR PDC FROM SACCHAROMYCES UVARUM IS NOT KNOWN, THE MODEL WAS BUILT ASSUMING THE ...SINCE THE SEQUENCE FOR PDC FROM SACCHAROMY | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.38 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 5.3 / Method: vapor diffusion, sitting drop | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.4 Å / Num. obs: 40681 / % possible obs: 92 % / Num. measured all: 192609 / Rmerge(I) obs: 0.069 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor Rwork: 0.197 / Rfactor obs: 0.197 / Highest resolution: 2.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2.4 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 10 Å / σ(I): 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
