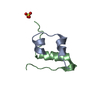
- PDB-1qkh: SOLUTION STRUCTURE OF THE RIBOSOMAL PROTEIN S19 FROM THERMUS THER... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 1qkh
Title
SOLUTION STRUCTURE OF THE RIBOSOMAL PROTEIN S19 FROM THERMUS THERMOPHILUS
Components
30S RIBOSOMAL PROTEIN S19
Keywords
RIBOSOMAL PROTEIN / RIBOSOME / THERMUS THERMOPHILUS / S19
Function / homology
Function and homology information
ribosomal small subunit assembly / small ribosomal subunit / rRNA binding / structural constituent of ribosome / translation / cytoplasm Similarity search - Function
30s Ribosomal Protein S19; Chain A / Ribosomal protein S19/S15 / Ribosomal protein S19, bacterial-type / Ribosomal protein S15/S19, conserved site / Ribosomal protein S19 signature. / Ribosomal protein S19/S15 / Ribosomal protein S19/S15, superfamily / Ribosomal protein S19 / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Method: simulated annealing / Software ordinal: 1 Details: THE STRUCTURE WAS DETERMINED USING 1104 DISTANCE RESTRAINTS, 42 DIHEDRAL ANGLE RESTRAINTS AND 14 HYDROGEN BOND RESTRAINTS. 50 STRUCTURES WERE CALCULATED AND REFINED USING AN AB INITIO ...Details: THE STRUCTURE WAS DETERMINED USING 1104 DISTANCE RESTRAINTS, 42 DIHEDRAL ANGLE RESTRAINTS AND 14 HYDROGEN BOND RESTRAINTS. 50 STRUCTURES WERE CALCULATED AND REFINED USING AN AB INITIO SIMULATED ANNEALING PROTOCOL FOR X- PLOR AND THEN REFINED IN TWO STEPS. AN R-6 AVERAGING PROTOCOL WAS USED FOR NON-STEREOSPECIFICALLY ASSIGNED PROTONS [1]. DURING THE SIMULATED ANNEALING STEP AND THE FIRST REFINEMENT STEP ONLY THE REPULSIVE PART OF THE VAN DER WAALS INTERACTION WAS INCLUDED. IN THE SECOND REFINEMENT STEP THE VAN DER WAALS INTERACTION WAS PARAMETERIZED USING A LENNARD-JONES POTENTIAL INCLUDING THE ATTRACTIVE PART. 21 STRUCTURES WERE SELECTED ON THE BASIS OF CUMULATIVE RMSD VALUES OF STRUCTURES, ORDERED AFTER OVERALL ENERGY, AND RAMACHANDRAN BEHAVIOR FOR REGIONS WITH LOW RESTRAINT DENSITIES. [1] BRUNGER, A. T., CLORE, G. M., GRONENBORN, A. M. & KARPLUS, M. (1986). THREE-DIMENSIONAL STRUCTURE OF PROTEINS DETERMINED BY MOLECULAR DYNAMICS WITH INTERPROTON DISTANCE RESTRAINTS: APPLICATION TO CRAMBIN. PROC NATL ACAD SCI USA 83, 3801-3805. OTHER DETAILS OF STRUCTURE REFINEMENT CAN BE FOUND IN THE JRNL CITATION.
NMR ensemble
Conformer selection criteria: CUMULATIVE RMSD OF STRUCTURES SORTED AFTER TOTAL ENERGY Conformers calculated total number: 50 / Conformers submitted total number: 21
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 THERMUS THERMOPHILUS (bacteria)
THERMUS THERMOPHILUS (bacteria) Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


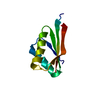






 PDBj
PDBj



 Assembly
Assembly
 HNCA
HNCA Sample preparation
Sample preparation Processing
Processing