 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1n1l: CRYSTAL STRUCTURE OF HCV NS3 PROTEASE DOMAIN:NS4A PEPTIDE COMPLEX... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n1l | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HCV NS3 PROTEASE DOMAIN:NS4A PEPTIDE COMPLEX WITH COVALENTLY BOUND INHIBITOR (GW472467X) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | VIRAL PROTEIN / SERINE PROTEASE / NONSTRUCTURAL PROTEINS / COFACTOR PEPTIDE / HELICASE / INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated suppression of host JAK-STAT cascade via inhibition of STAT activity / positive regulation of hexokinase activity / translocation of peptides or proteins into host cell cytoplasm / symbiont-mediated perturbation of host cellular process / Toll-like receptor 2 binding / viral capsid assembly / hepacivirin / adhesion receptor-mediated virion attachment to host cell / TBC/RABGAPs / host cell mitochondrial membrane ...symbiont-mediated suppression of host JAK-STAT cascade via inhibition of STAT activity / positive regulation of hexokinase activity / translocation of peptides or proteins into host cell cytoplasm / symbiont-mediated perturbation of host cellular process / Toll-like receptor 2 binding / viral capsid assembly / hepacivirin / adhesion receptor-mediated virion attachment to host cell / TBC/RABGAPs / host cell mitochondrial membrane / host cell lipid droplet / symbiont-mediated transformation of host cell / symbiont-mediated suppression of host TRAF-mediated signal transduction / positive regulation of cytokinesis / symbiont-mediated perturbation of host cell cycle G1/S transition checkpoint / viral process / negative regulation of protein secretion / symbiont-mediated suppression of host JAK-STAT cascade via inhibition of STAT1 activity / endoplasmic reticulum-Golgi intermediate compartment membrane / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / SH3 domain binding / virion component / kinase binding / nucleoside-triphosphate phosphatase / viral nucleocapsid / channel activity / monoatomic ion transmembrane transport / clathrin-dependent endocytosis of virus by host cell / entry receptor-mediated virion attachment to host cell / Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases / RNA helicase activity / host cell perinuclear region of cytoplasm / host cell endoplasmic reticulum membrane / RNA helicase / symbiont-mediated suppression of host type I interferon-mediated signaling pathway / ribonucleoprotein complex / serine-type endopeptidase activity / viral translational frameshifting / symbiont-mediated activation of host autophagy / RNA-directed RNA polymerase / cysteine-type endopeptidase activity / viral RNA genome replication / RNA-directed RNA polymerase activity / fusion of virus membrane with host endosome membrane / viral envelope / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / negative regulation of transcription by RNA polymerase II / ATP hydrolysis activity / proteolysis / RNA binding / zinc ion binding / ATP binding Similarity search - Function | ||||||
| Biological species |  Hepatitis C virus Hepatitis C virus | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Andrews, D.M. / Chaignot, H. / Coomber, B.A. / Good, A.C. / Hind, S.L. / Jones, P.S. / Mill, G. / Robinson, J.E. / Skarzynski, T. / Slater, M.J. / Somers, D.O.N. | ||||||
 Citation Citation | Journal: Org.Lett. / Year: 2002 Title: Pyrrolidine-5,5-trans-lactams. 2. The use of X-ray Crystal Structure Data in the Optimisation of P3 and P4 Substituents Authors: Andrews, D.M. / Chaignot, H. / Coomber, B.A. / Good, A.C. / Hind, S.L. / Johnson, M.R. / Jones, P.S. / Mill, G. / Robinson, J.E. / Skarzynski, T. / Slater, M.J. / Somers, D.O.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n1l.cif.gz | 85.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n1l.ent.gz | 63.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1n1l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n1/1n1lftp://data.pdbj.org/pub/pdb/validation_reports/n1/1n1l | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21044.975 Da / Num. of mol.: 2 / Fragment: Protease domain / Mutation: A164T Source method: isolated from a genetically manipulated source Source: (gene. exp.) Hepatitis C virus / Genus: Hepacivirus / Gene: H STRAIN / Production host:  #2: Protein/peptide | Mass: 2394.039 Da / Num. of mol.: 2 / Fragment: Residues 21-39 / Source method: obtained synthetically Details: The peptide was chemically synthesized. The sequence of the protein is naturally found in HEPATITIS C VIRUS type 1B. References: GenBank: 5748511, UniProt: O39914*PLUS #3: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-TRL / { | 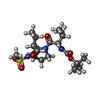  Mass: 433.563 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H35N3O6S Mass: 433.563 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H35N3O6S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.96 Å3/Da / Density % sol: 68.91 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: Inhibitor soaked into crystal generated according to Kim et al. (1996) Cell, 87, 343-355, pH 6.5, VAPOR DIFFUSION, SITTING DROP, temperature 277.0K |
| Crystal grow | *PLUS Details: Kim, J.L., (1996) 8, 344. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Details: MIRRORS |
| Radiation | Monochromator: YALE MIRRORS + Ni FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→40 Å / Num. all: 22723 / Num. obs: 21878 / % possible obs: 96.3 % / Observed criterion σ(I): 1 / Rmerge(I) obs: 0.052 / Net I/σ(I): 16.6 |
| Reflection shell | Resolution: 2.6→2.69 Å / Rmerge(I) obs: 0.238 / Mean I/σ(I) obs: 4.5 / Num. unique all: 2074 / % possible all: 92 |
| Reflection | *PLUS Lowest resolution: 40 Å |
| Reflection shell | *PLUS % possible obs: 92 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.6→20 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.931 / Cross valid method: THROUGHOUT / σ(F): 1 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.8 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 45.569 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.667 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 20 Å / Rfactor Rfree: 0.22 / Rfactor Rwork: 0.183 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
