 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mcv: Crystal Structure Analysis of a Hybrid Squash Inhibitor in Comple... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mcv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure Analysis of a Hybrid Squash Inhibitor in Complex with Porcine Pancreatic Elastase | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / elastase-inhibitor complex / hybrid squash inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationpancreatic elastase / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Ay, J. / Hilpert, K. / Krauss, N. / Schneider-Mergener, J. / Hoehne, W. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: Structure of a hybrid squash inhibitor in complex with porcine pancreatic elastase at 1.8 A resolution. Authors: Ay, J. / Hilpert, K. / Krauss, N. / Schneider-Mergener, J. / Hohne, W. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2000Title: Atomic Resolution Structure of native porcine pancreatic elastase at 1.1 A Authors: Wurtele, M. / Hahn, M. / Hilpert, K. / Hohne, W. #2: Journal: Embo J. / Year: 1986Title: X-ray crystal structure of the complex of human leukocyte elastase (PMN elastase) and the third DOMAIN OF the turkey ovomucoid inhibitor Authors: Bode, W. / Wei, A.Z. / Huber, R. / Meyer, E. / Travis, J. / Neumann, S. #3: Journal: FEBS Lett. / Year: 1989Title: THE REFINED 2.0 ANGSTROMS X-RAY CRYSTAL STRUCTURE OF THE COMPLEX FORMED BETWEEN BOVINE BETA-TRYPSIN AND CMTI-I, A TRYPSIN INHIBITOR FROM SQUASH SEEDS (CUCURBITA MAXIMA). TOPOLOGICAL SIMILARITY ...Title: THE REFINED 2.0 ANGSTROMS X-RAY CRYSTAL STRUCTURE OF THE COMPLEX FORMED BETWEEN BOVINE BETA-TRYPSIN AND CMTI-I, A TRYPSIN INHIBITOR FROM SQUASH SEEDS (CUCURBITA MAXIMA). TOPOLOGICAL SIMILARITY OF THE SQUASH SEED INHIBITORS WITH THE CARBOXYPEPTIDASE A INHIBITOR FROM POTATOES Authors: Bode, W. / Greyling, H.J. / Huber, R. / Otlewski, J. / Wilusz, T. | ||||||
| History |
| ||||||
| Remark 400 | COMPOUND RESIDUE NUMBERING FOR ELASTASE FOLLOWS THE NUMBERING OF BOVINE CHYMOTRYPSINOGEN A. |
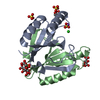
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mcv.cif.gz | 74.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mcv.ent.gz | 53.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1mcv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mc/1mcvftp://data.pdbj.org/pub/pdb/validation_reports/mc/1mcv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qnjS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25929.016 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Protein/peptide | Mass: 2973.452 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: The peptide was chemically synthesized using solid phase peptide synthesis. It was constructed using the trypsin binding loop of squash inhibitor EETI II, substituted by the sequence of a ...Details: The peptide was chemically synthesized using solid phase peptide synthesis. It was constructed using the trypsin binding loop of squash inhibitor EETI II, substituted by the sequence of a peptide that was derived from the third domain of the turkey ovomucoid inhibitor. Keywords: trypsin binding loop of squash inhibitor EETI II, substituted by the sequence of a peptide that was derived from the third domain of the turkey ovomucoid inhibitor |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38.51 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6 Details: sodium citrate, ammonium acetate, pH 6.0, VAPOR DIFFUSION, HANGING DROP, temperature 293.0K |
| Crystal grow | *PLUS Temperature: 293 K |
| Components of the solutions | *PLUS Conc.: 0.02 M / Common name: citrate / Details: pH6.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.9073 Å / Beamline: X11 / Wavelength: 0.9073 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Apr 4, 2002 |
| Radiation | Monochromator: triangular monochromator, bent mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9073 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→14 Å / Num. all: 22112 / Num. obs: 21938 / % possible obs: 98.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 3.6 % / Biso Wilson estimate: 13.5 Å2 / Rsym value: 0.037 / Net I/σ(I): 21 |
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 3.4 % / Mean I/σ(I) obs: 4.5 / Num. unique all: 1941 / Rsym value: 0.116 / % possible all: 87.8 |
| Reflection | *PLUS Num. obs: 22112 / Num. measured all: 79096 / Rmerge(I) obs: 0.037 |
| Reflection shell | *PLUS % possible obs: 87.8 % / Num. unique obs: 1941 / Rmerge(I) obs: 0.116 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QNJ Resolution: 1.8→14 Å / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||||
| Solvent computation | Solvent model: bulk solvent / Bsol: 40.2 Å2 / ksol: 0.32 e/Å3 | |||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.3 Å2
| |||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→14 Å
| |||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.86 Å
| |||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 14 Å / % reflection Rfree: 5 % | |||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
