 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1khq | ||||||
|---|---|---|---|---|---|---|---|
| Title | ORTHORHOMBIC FORM OF PAPAIN/ZLFG-DAM COVALENT COMPLEX | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / PROTEASE INHIBITOR / DIAZOMETHYLKETONE INHIBITOR / IRREVERSIBLE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
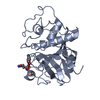
| Function / homology |  Function and homology information Function and homology informationpapain / serpin family protein binding / cysteine-type peptidase activity / proteolysis Similarity search - Function | ||||||
| Biological species |   Carica papaya (papaya) Carica papaya (papaya) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Janowski, R. / Kozak, M. / Jankowska, E. / Grzonka, Z. / Jaskolski, M. | ||||||
 Citation Citation | Journal: J.Pept.Res. / Year: 2004 Title: Two polymorphs of a covalent complex between papain and a diazomethylketone inhibitor Authors: Janowski, R. / Kozak, M. / Jankowska, E. / Grzonka, Z. / Jaskolski, M. #1: Journal: ACTA BIOCHIM.POL. / Year: 1997Title: Crystallization and preliminary crystallographic studies of a new crystal form of papain from Carica papaya. Authors: Kozak, M. / Kozian, E. / Grzonka, Z. / Jaskolski, M. #2: Journal: ACTA BIOCHIM.POL. / Year: 2001Title: Structural studies of cysteine proteases and their inhibitors Authors: Grzonka, Z. / Jankowska, E. / Wieczerzak, E. / Kasprzykowski, F. / Lankiewicz, L. / Wiczk, W. / Drabik, P. / Ciarkowski, J. / Janowski, R. / Kozak, M. / Jaskolski, M. / Grubb, A. #3: Journal: Biochemistry / Year: 1976Title: Binding of chloromethyl ketone substrate analogues to crystalline papain. Authors: Drenth, J. / Kalk, K.H. / Swen, H.M. #4: Journal: ACTA CRYSTALLOGR.,SECT.B / Year: 1992Title: Structure of monoclinic papain at 1.60 Angstroms resolution Authors: Pickersgill, R.W. / Harris, G.W. / Garman, E. #5: Journal: BIOCHIM.BIOPHYS.ACTA / Year: 1999Title: Binding modes of a new epoxysuccinyl-peptide inhibitor of cysteine proteases. Where and how do cysteine proteases express their selectivity? Authors: Czaplewski, C. / Grzonka, Z. / Jaskolski, M. / Kasprzykowski, F. / Kozak, M. / Politowska, E. / Ciarkowski, J. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN THE PEPTIDIC INHIBITOR IS COVALENTLY LINKED TO THE SG ATOM OF CYS 25 OF THE PROTEIN VIA A ...HETEROGEN THE PEPTIDIC INHIBITOR IS COVALENTLY LINKED TO THE SG ATOM OF CYS 25 OF THE PROTEIN VIA A METHYLKETONE GROUP OF THE HETGROUP GLM (-N-CH2-CO-CH2-). THE NON-STANDARD TRIPEPTIDE IS N-TERMINALLY PROTECTED BY THE BENZYLOXYCARBONYL- (Z- OR CBZ-) GROUP (NOT VISIBLE IN ELECTRON DENSITY). |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1khq.cif.gz | 58.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1khq.ent.gz | 42 KB | Display | PDB format |
| PDBx/mmJSON format | 1khq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kh/1khqftp://data.pdbj.org/pub/pdb/validation_reports/kh/1khq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1khpC  1ppnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23452.301 Da / Num. of mol.: 1 / Fragment: Papain, Residues 134-345 / Source method: isolated from a natural source / Source: (natural) Carica papaya (papaya) / References: UniProt: P00784, papain |
|---|---|
| #2: Protein/peptide | 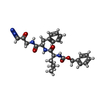  Type: Peptide-like / Class: Inhibitor / Mass: 512.001 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: L-leucyl-N-(2-oxopropyl)-L-phenylalaninamide Type: Peptide-like / Class: Inhibitor / Mass: 512.001 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: L-leucyl-N-(2-oxopropyl)-L-phenylalaninamide |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THE PEPTIDIC INHIBITOR IS COVALENTLY LINKED TO THE SG ATOM OF CYS 25 OF THE PROTEIN VIA A ...THE PEPTIDIC INHIBITOR IS COVALENTLY |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.12 Å3/Da / Density % sol: 42 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 9.2 Details: 72% methanol/ethanol (2:1), 34 mM NaCl, 50 mM 2-aminoethanol/HCl, pH 9.2, VAPOR DIFFUSION, HANGING DROP, temperature 292K | ||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 292 K / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 289 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: MACSCIENCE / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 22, 1999 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→20 Å / Num. all: 26000 / Num. obs: 26000 / % possible obs: 94.5 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 8.1 % / Biso Wilson estimate: 23.1 Å2 / Rmerge(I) obs: 0.059 / Net I/σ(I): 14.7 |
| Reflection shell | Resolution: 1.61→1.67 Å / Redundancy: 3 % / Rmerge(I) obs: 0.29 / Mean I/σ(I) obs: 3.8 / % possible all: 89.6 |
| Reflection | *PLUS Num. measured all: 211148 |
| Reflection shell | *PLUS Highest resolution: 1.63 Å / % possible obs: 89.6 % / Rmerge(I) obs: 0.29 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PPN Resolution: 1.6→10 Å / SU B: 1.91608 / SU ML: 0.06724 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.07936 / ESU R Free: 0.07645 / Stereochemistry target values: Engh & Huber Details: MAXIMUM LIKELIHOOD ALGORITHM. TLS PARAMETERS WERE USED. PARAMETERS WERE USED. THE CBZ (CARBOBENZOXY- OR BENZYLOXYCARBONYL-) BLOCKING GROUP AT THE N-TERMINUS OF THE INHIBITOR IS NOT VISIBLE ...Details: MAXIMUM LIKELIHOOD ALGORITHM. TLS PARAMETERS WERE USED. PARAMETERS WERE USED. THE CBZ (CARBOBENZOXY- OR BENZYLOXYCARBONYL-) BLOCKING GROUP AT THE N-TERMINUS OF THE INHIBITOR IS NOT VISIBLE IN ELECTRON DENSITY DUE TO DISORDER AND WAS NOT INCLUDED IN THE MODEL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.149 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
