 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kc7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Pyruvate Phosphate Dikinase with Bound Mg-phosphonopyruvate | ||||||
 Components Components | pyruvate phosphate dikinase | ||||||
 Keywords Keywords | TRANSFERASE / PHOSPHOTRANSFERASE / KINASE / phosphonopyruvate inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationpyruvate, phosphate dikinase / pyruvate, phosphate dikinase activity / kinase activity / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Clostridium symbiosum (bacteria) Clostridium symbiosum (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.2 Å | ||||||
 Authors Authors | Chen, C.C. / Howard, A. / Herzberg, O. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Pyruvate site of pyruvate phosphate dikinase: crystal structure of the enzyme-phosphonopyruvate complex, and mutant analysis Authors: Herzberg, O. / Chen, C.C. / Liu, S. / Tempczyk, A. / Howard, A. / Wei, M. / Ye, D. / Dunaway-Mariano, D. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1996Title: SWIVELING-DOMAIN MECHANISM FOR ENZYMATIC PHOSPHOTRANSFER BETWEEN REMOTE REACTION SITES Authors: Herzberg, O. / Chen, C.C.H. / Kapadia, G. / Mcguire, M. / Carroll, L.J. / Noh, S.J. / Dunaway-Mariano, D. #2: Journal: Biochemistry / Year: 1998Title: Location of the Phosphate Binding Site within Clostridium Symbiosum Pyruvate Phosphate dikinase Authors: Mcguire, M. / Huang, K. / Kapadia, G. / Herzberg, O. / Dunaway-Mariano, D. #3: Journal: J.Biol.Chem. / Year: 2000Title: Identification of Domain-Domain Docking Sites within Clostridium Symbiosum Pyruvate Phosphate Dikinase by Amino Acid Replacement Authors: Wei, M. / Li, Z. / Ye, D. / Herzberg, O. / Dunaway-Mariano, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kc7.cif.gz | 180.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kc7.ent.gz | 141.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1kc7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kc/1kc7ftp://data.pdbj.org/pub/pdb/validation_reports/kc/1kc7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kblC  1dikS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | A dimer generated by a crystallographic 2-fold axis: -1.0 0.0 0.0 89.837 0.0 1.0 0.0 0.0 0.0 0.0 -1.0 0.0 |
-Components
| #1: Protein | Mass: 96640.148 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Clostridium symbiosum (bacteria) / Gene: PPDK / Plasmid: PACYC184-D12 / Production host: | ||
|---|---|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-PPR / | 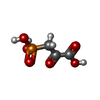  Mass: 168.042 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5O6P Mass: 168.042 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5O6P |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.75 Å3/Da / Density % sol: 55.33 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 303 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 54% saturated ammonium sulfate, 100mM Hepes buffer (pH 7.0), 9mg/ml protein solution consisting of: 20mM imidazole buffer (pH 6.5), 100mM KCl, 40mM phosphonopyruvate, 5mM MgCl2, 0.1mM EDTA, ...Details: 54% saturated ammonium sulfate, 100mM Hepes buffer (pH 7.0), 9mg/ml protein solution consisting of: 20mM imidazole buffer (pH 6.5), 100mM KCl, 40mM phosphonopyruvate, 5mM MgCl2, 0.1mM EDTA, 1mM DTT, VAPOR DIFFUSION, HANGING DROP, temperature 303K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 30 ℃ / pH: 6.5 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 6, 2001 / Details: mirror |
| Radiation | Monochromator: CRYOGENITCALLY-COOLED SI(111) DOUBLE CRYSTAL system Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.73→25 Å / Num. all: 104444 / Num. obs: 104444 / % possible obs: 95.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.4 % / Biso Wilson estimate: 40.8 Å2 / Rmerge(I) obs: 0.114 / Net I/σ(I): 26.6 |
| Reflection shell | Resolution: 1.73→1.79 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 4.1 / Num. unique all: 14814 / % possible all: 81.4 |
| Reflection | *PLUS |
| Reflection shell | *PLUS % possible obs: 81.4 % / Num. unique obs: 14814 / Rmerge(I) obs: 0.523 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 1DIK Resolution: 2.2→25 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→25 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 25 Å / Num. reflection obs: 49319 / σ(F): 2 / % reflection Rfree: 4.8 % / Rfactor obs: 0.198 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: c_angle_deg / Dev ideal: 1.9 |
