 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1k3t: Structure of Glycosomal Glyceraldehyde-3-Phosphate Dehydrogenase ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1k3t | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Glycosomal Glyceraldehyde-3-Phosphate Dehydrogenase from Trypanosoma cruzi Complexed with Chalepin, a Coumarin Derivative Inhibitor | ||||||
 Components Components | Glyceraldehyde-3-phosphate dehydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Apo-Protein / gGAPDH-Chalepin complex / glycosome / Trypanosoma cruzi | ||||||
| Function / homology |  Function and homology information Function and homology informationglyceraldehyde-3-phosphate dehydrogenase (phosphorylating) / glycosome / glyceraldehyde-3-phosphate dehydrogenase (NAD+) (phosphorylating) activity / glycolytic process / glucose metabolic process / NAD binding / NADP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Pavao, F. | ||||||
 Citation Citation | Journal: FEBS Lett. / Year: 2002 Title: Structure of Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase complexed with chalepin, a natural product inhibitor, at 1.95 A resolution. Authors: Pavao, F. / Castilho, M.S. / Pupo, M.T. / Dias, R.L. / Correa, A.G. / Fernandes, J.B. / da Silva, M.F. / Mafezoli, J. / Vieira, P.C. / Oliva, G. #1: Journal: Thesis / Year: 2001Title: Estudos Cristalograficos e Planejamento Racional de Inibidores Especificos da Enzima Gliceraldeido-3-fosfato Desidrogenase Glicossomal (gGAPDH) de Trypanosoma cruzi Authors: Pavao, F. #2: Journal: Pure Appl.Chem. / Year: 2001Title: Strategies for the Isolation and Identification of Trypanocidal Compounds from the Rutales Authors: Vieira, P.C. / Mafezoli, J. / Pupo, M.T. / Fernandes, J.B. / Silva, M.F.G.F. / Albuquerque, S. / Oliva, G. / Pavao, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1k3t.cif.gz | 299.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1k3t.ent.gz | 241 KB | Display | PDB format |
| PDBx/mmJSON format | 1k3t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k3/1k3tftp://data.pdbj.org/pub/pdb/validation_reports/k3/1k3t | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39112.539 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P22513, glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) #2: Chemical | ChemComp-BRZ / | 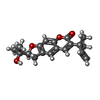  Mass: 314.376 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H22O4 Mass: 314.376 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H22O4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 937 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 937 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.338 Å3/Da / Density % sol: 47.45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.1 Details: 16% PEG 8000, 0.1M calcium acetate, 0.1M sodium cacodylate, pH 7.1, VAPOR DIFFUSION, HANGING DROP, temperature 295K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 7.6 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Jul 30, 1998 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→44.72 Å / Num. all: 81630 / Num. obs: 81630 / % possible obs: 77.9 % / Redundancy: 2.4 % / Biso Wilson estimate: 22.48 Å2 / Rmerge(I) obs: 0.107 / Net I/σ(I): 6.78 |
| Reflection shell | Resolution: 1.95→2.02 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.483 / Mean I/σ(I) obs: 1.1 / Num. unique all: 3980 / % possible all: 38.1 |
| Reflection | *PLUS Num. measured all: 195798 / Rmerge(I) obs: 0.107 |
| Reflection shell | *PLUS % possible obs: 38.1 % / Rmerge(I) obs: 0.483 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Holo-D-Glyceraldehyde-3-phosphate dehydrogenase from T. cruzi solved at 2.8 A resolution by Souza et al., FEBS Letters, 424 (1998), 131-135. Resolution: 1.95→15 Å / SU B: 6.158 / SU ML: 0.172 / Cross valid method: THROUGHOUT / ESU R: 0.267 / ESU R Free: 0.233 Stereochemistry target values: John Priestle's with added co-factors from York. Details: REFINEMENT PERFORMED WITHOUT NON-CRYSTALLOGRAPHIC SYMMETRY. FROM THE INITIAL CYCLES OF THE REFINEMENT, IT WAS EVIDENT THAT THE INHIBITOR WAS CLOSELY PACKED AGAINST THE ACTIVE SITE RESIDUE ...Details: REFINEMENT PERFORMED WITHOUT NON-CRYSTALLOGRAPHIC SYMMETRY. FROM THE INITIAL CYCLES OF THE REFINEMENT, IT WAS EVIDENT THAT THE INHIBITOR WAS CLOSELY PACKED AGAINST THE ACTIVE SITE RESIDUE CYS166, INDUCING A CONFORMATIONAL CHANGE OF THE -SH GROUP OF THIS AMINOACID THROUGH A ROTATION OF c1 (Chi1 angle), IN ORDER TO ACCOMMODATE THE POSITIONING OF THE INHIBITOR AT THE ACTIVE SITE. THE FINAL ELECTRON DENSITY MAPS SHOW TWO ALTERNATIVE CONFORMATIONS OF CYS166, ONE OF THEM APPARENTLY CONNECTING TO THE INHIBITOR. THIS IS AN ARTIFACT, SINCE THE SIDE CHAIN OF THIS RESIDUE CAN ONLY BE AT THE NATIVE CONFORMATION IF THE INHIBITOR IS ABSENT. THEREFORE, THE EVIDENT TWO CONFORMATIONS OF THE CYS166 SIDE CHAIN GUARANTEES THAT THE INHIBITOR HAS TO HAVE PARTIAL OCCUPANCY. THE FIRST 160 RESIDUES FROM N-TERM AND THE LAST 7 RESIDUES FROM C-TERM OF MONOMER C SHOWS HIGH DEGREE OF STATIC DISORDER DUE TO PARTIAL OCCUPANCY OF INHIBITOR (85%), RESULTING IN A LACK OF ELECTRON DENSITY TO THE SEVERAL SIDE CHAINS AND TO SOME BACKBONE ATOMS. IN SPITE OF THIS, THE COORDINATES OF RESPECTIVES SIDE CHAINS ARE KEPT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.81 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 15 Å / % reflection Rfree: 3 % / Rfactor Rfree: 0.28 / Rfactor Rwork: 0.201 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
