 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jan: COMPLEX OF PRO-LEU-GLY-HYDROXYLAMINE WITH THE CATALYTIC DOMAIN OF... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jan | ||||||
|---|---|---|---|---|---|---|---|
| Title | COMPLEX OF PRO-LEU-GLY-HYDROXYLAMINE WITH THE CATALYTIC DOMAIN OF MATRIX METALLO PROTEINASE-8 (PHE79 FORM) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / METALLOPROTEASE / ZINC-ENDOPEPTIDASE / METZINCINS / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationneutrophil collagenase / tumor necrosis factor binding / positive regulation of microglial cell activation / positive regulation of tumor necrosis factor-mediated signaling pathway / endodermal cell differentiation / positive regulation of neuroinflammatory response / Activation of Matrix Metalloproteinases / Collagen degradation / collagen catabolic process / extracellular matrix disassembly ...neutrophil collagenase / tumor necrosis factor binding / positive regulation of microglial cell activation / positive regulation of tumor necrosis factor-mediated signaling pathway / endodermal cell differentiation / positive regulation of neuroinflammatory response / Activation of Matrix Metalloproteinases / Collagen degradation / collagen catabolic process / extracellular matrix disassembly / Degradation of the extracellular matrix / extracellular matrix organization / metalloendopeptidase activity / specific granule lumen / positive regulation of tumor necrosis factor production / tertiary granule lumen / peptidase activity / cellular response to lipopolysaccharide / extracellular matrix / endopeptidase activity / serine-type endopeptidase activity / Neutrophil degranulation / proteolysis / : / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.5 Å X-RAY DIFFRACTION / Resolution: 2.5 Å | ||||||
 Authors Authors | Reinemer, P. / Grams, F. / Huber, R. / Kleine, T. / Schnierer, S. / Pieper, M. / Tschesche, H. / Bode, W. | ||||||
 Citation Citation | Journal: FEBS Lett. / Year: 1994 Title: Structural implications for the role of the N terminus in the 'superactivation' of collagenases. A crystallographic study. Authors: Reinemer, P. / Grams, F. / Huber, R. / Kleine, T. / Schnierer, S. / Piper, M. / Tschesche, H. / Bode, W. #1: Journal: Eur.J.Biochem. / Year: 1995Title: X-Ray Structures of Human Neutrophil Collagenase Complexed with Peptide Hydroxamate and Peptide Thiol Inhibitors. Implications for Substrate Binding and Rational Drug Design Authors: Grams, F. / Reinemer, P. / Powers, J.C. / Kleine, T. / Pieper, M. / Tschesche, H. / Huber, R. / Bode, W. #2: Journal: Embo J. / Year: 1994Title: The X-Ray Crystal Structure of the Catalytic Domain of Human Neutrophil Collagenase Inhibited by a Substrate Analogue Reveals the Essentials for Catalysis and Specificity Authors: Bode, W. / Reinemer, P. / Huber, R. / Kleine, T. / Schnierer, S. / Tschesche, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jan.cif.gz | 58.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jan.ent.gz | 42.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1jan.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ja/1janftp://data.pdbj.org/pub/pdb/validation_reports/ja/1jan | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 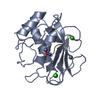
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18258.918 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, RESIDUES 79 - 242 Source method: isolated from a genetically manipulated source Details: MMP-8 IS IDENTICAL TO THE HUMAN NEUTROPHIL COLLAGENASE Source: (gene. exp.) Homo sapiens (human) / Cell: NEUTROPHILS / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | 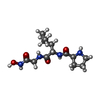  Type: Peptide-like / Class: Inhibitor / Mass: 300.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: L-prolyl-L-leucyl-N-hydroxyglycinamide Type: Peptide-like / Class: Inhibitor / Mass: 300.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: L-prolyl-L-leucyl-N-hydroxyglycinamide | ||||
| #3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 41 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 6 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|