 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1j4r | ||||||
|---|---|---|---|---|---|---|---|
| Title | FK506 BINDING PROTEIN COMPLEXED WITH FKB-001 | ||||||
 Components Components | FK506-BINDING PROTEIN | ||||||
 Keywords Keywords | ISOMERASE / ROTAMASE / INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationmacrolide binding / activin receptor binding / regulation of skeletal muscle contraction by regulation of release of sequestered calcium ion / cytoplasmic side of membrane / transforming growth factor beta receptor binding / TGFBR1 LBD Mutants in Cancer / type I transforming growth factor beta receptor binding / negative regulation of activin receptor signaling pathway / signaling receptor inhibitor activity / heart trabecula formation ...macrolide binding / activin receptor binding / regulation of skeletal muscle contraction by regulation of release of sequestered calcium ion / cytoplasmic side of membrane / transforming growth factor beta receptor binding / TGFBR1 LBD Mutants in Cancer / type I transforming growth factor beta receptor binding / negative regulation of activin receptor signaling pathway / signaling receptor inhibitor activity / heart trabecula formation / I-SMAD binding / regulation of amyloid precursor protein catabolic process / terminal cisterna / ryanodine receptor complex / 'de novo' protein folding / ventricular cardiac muscle tissue morphogenesis / FK506 binding / TGF-beta receptor signaling activates SMADs / mTORC1-mediated signalling / Calcineurin activates NFAT / regulation of immune response / heart morphogenesis / supramolecular fiber organization / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / sarcoplasmic reticulum membrane / T cell activation / peptidylprolyl isomerase / sarcoplasmic reticulum / peptidyl-prolyl cis-trans isomerase activity / TGF-beta receptor signaling in EMT (epithelial to mesenchymal transition) / calcium channel regulator activity / negative regulation of transforming growth factor beta receptor signaling pathway / protein maturation / Z disc / SARS-CoV-1 activates/modulates innate immune responses / regulation of protein localization / protein refolding / protein folding / amyloid fibril formation / Potential therapeutics for SARS / transmembrane transporter binding / positive regulation of canonical NF-kappaB signal transduction / membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Sheriff, S. | ||||||
 Citation Citation | Journal: Org.Lett. / Year: 2001 Title: 2-Aryl-2,2-difluoroacetamide FKBP12 ligands: synthesis and X-ray structural studies. Authors: Dubowchik, G.M. / Vrudhula, V.M. / Dasgupta, B. / Ditta, J. / Chen, T. / Sheriff, S. / Sipman, K. / Witmer, M. / Tredup, J. / Vyas, D.M. / Verdoorn, T.A. / Bollini, S. / Vinitsky, A. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1, 2, 3 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 3 ... BIOMOLECULE: 1, 2, 3 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 3 CHAIN(S). SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). ALTHOUGH THE THREE MOLECULES ARE RELATED TO ONE ANOTHER IN THE ASYMMETRIC UNIT BY NON-CRYSTALLOGRAPHIC 3-FOLD SYMMETRY, THE BIOLOGICAL UNIT IS A MONOMER. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1j4r.cif.gz | 81.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1j4r.ent.gz | 60.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1j4r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j4/1j4rftp://data.pdbj.org/pub/pdb/validation_reports/j4/1j4r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bl4S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11836.508 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  #2: Chemical | 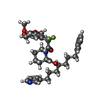  Mass: 624.715 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C35H42F2N2O6 Mass: 624.715 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C35H42F2N2O6#3: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 39 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 2.5 M AMMONIUM SULFATE, 0.1 M HEPES, PH 7.5, CRYOPROTECTANT 20% (V/V) GLYCEROL | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: BRUKER / Detector: AREA DETECTOR / Date: May 22, 1998 / Details: SUPPER 60MM MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.79→15 Å / Num. obs: 25481 / % possible obs: 98.5 % / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Biso Wilson estimate: 8.2 Å2 / Rmerge(I) obs: 0.055 / Net I/σ(I): 18.8 |
| Reflection shell | Resolution: 1.79→1.9 Å / Redundancy: 2.2 % / Rmerge(I) obs: 0.168 / Mean I/σ(I) obs: 4.4 / % possible all: 92.2 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 9999 Å / % possible obs: 98 % / Num. measured all: 77913 / Rmerge(I) obs: 0.042 |
| Reflection shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.9 Å / % possible obs: 92 % / Num. unique obs: 3970 / Num. measured obs: 8852 / Rmerge(I) obs: 0.155 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: FKBP FROM PDB ENTRY 1BL4 Resolution: 1.8→8 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.91 Å / Rfactor Rfree error: 0.025 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 1 / % reflection Rfree: 3.9 % / Rfactor Rfree: 0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 13.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.289 / % reflection Rfree: 3.7 % / Rfactor Rwork: 0.235 |
