 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hq2: CRYSTAL STRUCTURE OF A TERNARY COMPLEX OF E.COLI HPPK(R82A) WITH ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hq2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A TERNARY COMPLEX OF E.COLI HPPK(R82A) WITH MGAMPCPP AND 6-HYDROXYMETHYL-7,8-DIHYDROPTERIN AT 1.25 ANGSTROM RESOLUTION | ||||||
 Components Components | 6-HYDROXYMETHYL-7,8-DIHYDROPTERIN PYROPHOSPHOKINASE | ||||||
 Keywords Keywords | TRANSFERASE / PYROPHOSPHOKINASE / PYROPHOSPHORYL TRANSFER / CATALYTIC MECHANISM / FOLATE / HPPK / PTERIN / 6-HYDROXYMETHYL-7 / 8-DIHYDROPTERIN / TERNARY COMPLEX / SUBSTRATE SPECIFICITY / ANTIMICROBIAL AGENT / DRUG DESIGN | ||||||
| Function / homology |  Function and homology information Function and homology information2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase / 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase activity / folic acid biosynthetic process / tetrahydrofolate biosynthetic process / kinase activity / magnesium ion binding / ATP binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.25 Å | ||||||
 Authors Authors | Blaszczyk, J. / Ji, X. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Dynamic Roles of Arginine Residues 82 and 92 of Escherichia coli 6-Hydroxymethyl-7,8-dihydropterin Pyrophosphokinase: Crystallographic Studies Authors: Blaszczyk, J. / Li, Y. / Shi, G. / Yan, H. / Ji, X. #1: Journal: Structure / Year: 1999Title: Crystal Structure of 6-Hydroxymethyl-7,8-Dihydropterin Pyrophosphokinase, a Potential Target for the Development of Novel Antimicrobial Agents Authors: Xiao, B. / Shi, G. / Chen, X. / Yan, H. / Ji, X. #2: Journal: Structure / Year: 2000Title: Catalytic Center Assembly of HPPK as Revealed by the Crystal Structure of a Ternary Complex at 1.25 A Resolution Authors: Blaszczyk, J. / Shi, G. / Yan, H. / Ji, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hq2.cif.gz | 97.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hq2.ent.gz | 71.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1hq2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hq/1hq2ftp://data.pdbj.org/pub/pdb/validation_reports/hq/1hq2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1f9hC  1g4cC  1im6C  1kbrC  3ip0C  1eqo 1f9y C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 17880.418 Da / Num. of mol.: 1 / Mutation: R82A Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P26281, 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase |
|---|
-Non-polymers , 6 types, 341 molecules 


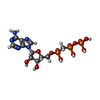


| #2: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-CL / |  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-ACT /  Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2#5: Chemical | ChemComp-APC / |  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM#6: Chemical | ChemComp-PH2 / |  Mass: 195.179 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H9N5O2 Mass: 195.179 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H9N5O2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 332 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.92 Å3/Da / Density % sol: 33.45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: PEG4000, MAGNESIUM CHLORIDE, ACETATE, GLYCEROL , pH 4.6, VAPOR DIFFUSION, HANGING DROP, temperature 292.0K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18-20 ℃ / pH: 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X9B / Wavelength: 0.96394 / Wavelength: 0.96394 Å / Beamline: X9B / Wavelength: 0.96394 / Wavelength: 0.96394 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Nov 12, 2000 / Details: MIRROR |
| Radiation | Monochromator: SILICON 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.96394 Å / Relative weight: 1 |
| Reflection | Resolution: 1.25→30 Å / Num. all: 39345 / Num. obs: 39345 / % possible obs: 99.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.211 % / Biso Wilson estimate: 12.3 Å2 / Rmerge(I) obs: 0.092 / Net I/σ(I): 12.2875 |
| Reflection shell | Resolution: 1.25→1.29 Å / Redundancy: 3.079 % / Rmerge(I) obs: 0.571 / Mean I/σ(I) obs: 1.678 / Num. unique all: 3863 / % possible all: 99 |
| Reflection | *PLUS % possible obs: 99.3 % / Num. measured all: 126338 |
| Reflection shell | *PLUS % possible obs: 99 % / Mean I/σ(I) obs: 1.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1EQO 1eqo Resolution: 1.25→30 Å / Num. parameters: 14475 / Num. restraintsaints: 16997 / Isotropic thermal model: ANISOTROPIC / Cross valid method: FREE R / σ(F): 4 / σ(I): 2 / Stereochemistry target values: ENGH AND HUBER Details: FULL MATRIX LEAST-SQUARES PROCEDURE, WITH BLOCK OF PARAMETERS SET FOR EACH CYCLE OF ANISOTROPIC REFINEMENT
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL. 91 (1975) 201-228 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.307 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.096 Å / Luzzati d res low obs: 5 Å / Num. disordered residues: 9 / Occupancy sum hydrogen: 1297 / Occupancy sum non hydrogen: 1644.5 | ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.25→30 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.172 / Rfactor Rwork: 0.134 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
