SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Compound details
THE PROTEIN (CHAIN A) WAS CRYSTALLIZED IN THE PRESENCE OF A PEPTIDE FRAGEMENT FROM ENDOTHELIAL PAS ...THE PROTEIN (CHAIN A) WAS CRYSTALLIZED IN THE PRESENCE OF A PEPTIDE FRAGEMENT FROM ENDOTHELIAL PAS DOMAIN PROTEIN 1 SWISS-PROT ID Q99814 (RESIDUES 846-858) BUT NONE OF THE RESIDUES CORRESPONDING TO THE PEPTIDE WERE VISIBLE IN THE ELECTRON DENSITY MAPS. IT IS POSSIBLE THAT THE PEPTIDE DID NOT BIND TO THE PROTEIN AND HENCE HAS NOT BEEN INCLUDED IN THE COMPND, SOURCE AND SEQRES RECORDS. THE SEQUENCE OF THE FRAGMENT IS GIVEN BELOW. VAL ASN VAL PRO VAL LEU GLY SER SER THR LEU LEU GLN
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.4 Å3/Da / Density % sol: 63 % / Description: SEE REMARK 400
Crystal grow
pH: 7.5 Details: 1.2M AMMONIUM SULPHATE, 4% PEG400, 0.1M HEPES PH7.5 ARGON ATMOSPHERE, 11MG/ML PROTEIN WITH 1MM FE(II), 2.5MM AKG AND 2.5MM PEPTIDE (SEE REMARK 400), pH 7.50
Crystal grow
*PLUS
Temperature: 17 ℃ / Method: vapor diffusion, hanging drop
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






















 PDBj
PDBj






 Assembly
Assembly



 Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe
Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe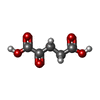
 Mass: 146.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6O5
Mass: 146.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6O5
 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PX9.5 / Wavelength: 0.92
/ Beamline: PX9.5 / Wavelength: 0.92  Processing
Processing