 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ao0: GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE (PRPP) AMIDOTRANSFERASE FRO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ao0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE (PRPP) AMIDOTRANSFERASE FROM B. SUBTILIS COMPLEXED WITH ADP AND GMP | ||||||
 Components Components | GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE AMIDOTRANSFERASE | ||||||
 Keywords Keywords | GLUTAMINE AMIDOTRANSFERASE / TRANSFERASE / PRTASE / PURINE BIOSYNTHESIS / PHOSPHORIBOSYLTRANSFERASE / GLYCOSYLTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationamidophosphoribosyltransferase / amidophosphoribosyltransferase activity / purine nucleobase biosynthetic process / purine nucleotide biosynthetic process / 'de novo' IMP biosynthetic process / 4 iron, 4 sulfur cluster binding / magnesium ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Tomchick, D.R. / Smith, J.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Authors: Chen, S. / Tomchick, D.R. / Wolle, D. / Hu, P. / Smith, J.L. / Switzer, R.L. / Zalkin, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ao0.cif.gz | 359.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ao0.ent.gz | 293 KB | Display | PDB format |
| PDBx/mmJSON format | 1ao0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ao/1ao0ftp://data.pdbj.org/pub/pdb/validation_reports/ao/1ao0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gphS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||
| Details | THE AMIDOTRANSFERASE MOLECULE IS A HOMOTETRAMER IN THE CRYSTALLINE STATE, BUT THE AGGREGATION STATE IN SOLUTION IS CONCENTRATION DEPENDENT. CRYSTALS CONTAIN 1 TETRAMER PER ASYMMETRIC UNIT AND COORDINATES FOR THE ENTIRE TETRAMER ARE INCLUDED IN THE ENTRY. THE SUBUNITS HAVE CHAIN IDENTIFIERS A, B, C, AND D. EACH "CHAIN" INCLUDES 455 AMINO ACID RESIDUES, ONE [4FE-4S] CLUSTER (RESIDUE 466), ONE GMP MOLECULE (RESIDUE 467), ONE ADP MOLECULE (RESIDUE 468), ONE MAGNESIUM ION (RESIDUE 469) AND ONE WATER MOLECULE LIGATED TO THE MAGNESIUM (CHAIN IDENTIFIER S). MOLECULAR P, Q AND R AXES RELATING SUBUNITS B, D, AND C, RESPECTIVELY, TO SUBUNIT A ARE SPECIFIED IN THE MTRIX RECORDS. THE FIRST MATRIX PRESENTED IN *MTRIX* RECORDS BELOW IS A MOLECULAR P AXIS MATRIX. THE SECOND IS A MOLECULAR R AXIS MATRIX, AND THE THIRD IS A MOLECULAR Q AXIS MATRIX. |
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 49878.469 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|
-Non-polymers , 5 types, 20 molecules 

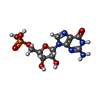


| #2: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-SF4 /  Mass: 351.640 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 351.640 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe4S4#4: Chemical | ChemComp-5GP /  Mass: 363.221 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H14N5O8P Mass: 363.221 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H14N5O8P#5: Chemical | ChemComp-ADP /  Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | PRO 86 IN ALL CHAINS IS A CIS-PROLINE. ASP 282 IN ALL CHAINS IS A CIS-ASPARTATE, PART OF A ...PRO 86 IN ALL CHAINS IS A CIS-PROLINE. ASP 282 IN ALL CHAINS IS A CIS-ASPARTATE, PART OF A PHOSPHATE BINDING SITE INVOLVED IN BINDING BOTH SUBSTRATE AND INHIBITORS |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 40 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.9 Details: 24% PEG8000, 200MM KCL, 50MM EPPS PH 7.9,2 MM ADP, 2 MM GMP, 10 MM MGCL2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: microdialysis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Jan 1, 1995 |
| Radiation | Monochromator: MSC DOUBLE MIRROR / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→15 Å / Num. obs: 46771 / % possible obs: 91 % / Observed criterion σ(I): -3 / Redundancy: 4.2 % / Biso Wilson estimate: 47 Å2 / Rsym value: 0.08 / Net I/σ(I): 20.2 |
| Reflection | *PLUS Num. measured all: 195956 / Rmerge(I) obs: 0.08 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GPH Resolution: 2.8→15 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.88 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
