 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ecf: ESCHERICHIA COLI GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE (PRPP) AMI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ecf | ||||||
|---|---|---|---|---|---|---|---|
| Title | ESCHERICHIA COLI GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE (PRPP) AMIDOTRANSFERASE | ||||||
 Components Components | GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE AMIDOTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE (GLUTAMINE AMIDOTRANSFERASE) / PURINE BIOSYNTHESIS / TRANSFERASE / GLYCOSYLTRANSFERASE / GLUTAMINE AMIDOTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationamidophosphoribosyltransferase / amidophosphoribosyltransferase activity / purine nucleobase biosynthetic process / L-glutamine metabolic process / purine nucleotide biosynthetic process / 'de novo' IMP biosynthetic process / guanosine tetraphosphate binding / glycosyltransferase activity / magnesium ion binding / identical protein binding ...amidophosphoribosyltransferase / amidophosphoribosyltransferase activity / purine nucleobase biosynthetic process / L-glutamine metabolic process / purine nucleotide biosynthetic process / 'de novo' IMP biosynthetic process / guanosine tetraphosphate binding / glycosyltransferase activity / magnesium ion binding / identical protein binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT, SIR PHASES FROM SEMET PROTEIN / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT, SIR PHASES FROM SEMET PROTEIN / Resolution: 2 Å | ||||||
 Authors Authors | Krahn, J.M. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1998 Title: Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Authors: Muchmore, C.R. / Krahn, J.M. / Kim, J.H. / Zalkin, H. / Smith, J.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ecf.cif.gz | 233.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ecf.ent.gz | 186.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ecf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ec/1ecfftp://data.pdbj.org/pub/pdb/validation_reports/ec/1ecf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ecjC  1gphS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.0809, -0.0094, 0.9967), Vector: Details | PHE B 88 - SER B 92 EXISTS IN TWO DISTINCT CONFORMATIONS AS A RESULT OF THE BINDING OF PIPES BUFFER ALONG THE CRYSTALLOGRAPHIC MOLECULAR TWO-FOLD AXIS, AND IS THEREFORE POORLY DEFINED. APPLICATION OF THE CRYSTALLOGRAPHIC SYMMETRY OPERATOR TO THE SECOND SET OF RESIDUES WILL GENERATE A COMPLETE ASYMMETRIC MODEL OF THIS SITE. | |
-Components
| #1: Protein | Mass: 56422.684 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P00496, UniProt: P0AG16*PLUS, amidophosphoribosyltransferase #2: Chemical | ChemComp-PIN / 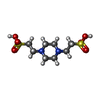  Mass: 302.368 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H18N2O6S2 / Comment: pH buffer*YM Mass: 302.368 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H18N2O6S2 / Comment: pH buffer*YM#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 986 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 986 / Source method: isolated from a natural source / Formula: H2OCompound details | GLU A 303 AND GLU B 303 ARE CIS-GLUTAMINE - PART OF A PHOSPHATE BINDING SITE INVOLVED IN BINDING ...GLU A 303 AND GLU B 303 ARE CIS-GLUTAMINE - PART OF A PHOSPHATE BINDING SITE INVOLVED IN BINDING BOTH SUBSTRATE AND INHIBITORS | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 40 % Description: ORIGINAL MOLECULAR REPLACEMENT SOLUTION WAS OBTAINED IN A DIFFERENT CRYSTAL FORM. | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.6 / Details: pH 5.6 | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6.5 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Mar 18, 1995 |
| Radiation | Monochromator: MSC DOUBLE MIRROR / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. obs: 65480 / % possible obs: 98.2 % / Observed criterion σ(I): -3 / Redundancy: 4.1 % / Rmerge(I) obs: 0.041 / Net I/σ(I): 24.1 |
| Reflection shell | Resolution: 2→2.03 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.225 / Mean I/σ(I) obs: 4.5 / % possible all: 92.7 |
| Reflection | *PLUS Num. measured all: 268969 |
| Reflection shell | *PLUS % possible obs: 92.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT, SIR PHASES FROM SEMET PROTEIN Starting model: 1GPH Resolution: 2→30 Å / σ(F): 0 Details: TYR 94 IN EACH CHAIN HAS PHI/PSI VALUES THAT ARE NORMALLY DISALLOWED. IT FORMS A SHARP BETA-TURN WITH PRO 93, A CIS-PROLINE. THE DISTANCE BETWEEN X982 AND X230 IS ARTIFICIALLY SHORT DUE TO ...Details: TYR 94 IN EACH CHAIN HAS PHI/PSI VALUES THAT ARE NORMALLY DISALLOWED. IT FORMS A SHARP BETA-TURN WITH PRO 93, A CIS-PROLINE. THE DISTANCE BETWEEN X982 AND X230 IS ARTIFICIALLY SHORT DUE TO UNMODELLED HETEROGENEITY AT A SPECIAL POSITION NEAR X230. ADDITIONAL UNMODELED ELECTRON DENSITY IS PRESENT NEAR THE CYS 1 SULFUR. IT APPEARS TO REPRESENT A HETEROGENEOUS OXIDATION BY OXYGEN AND SULFUR COMPOUNDS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.21 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 65453 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 28.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
