 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1914 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SIGNAL RECOGNITION PARTICLE ALU RNA BINDING HETERODIMER, SRP9/14 | ||||||
 Components Components | SIGNAL RECOGNITION PARTICLE 9/14 FUSION PROTEIN | ||||||
 Keywords Keywords | ALU DOMAIN / RNA BINDING / SIGNAL RECOGNITION PARTICLE (SRP) / TRANSLATION REGULATION | ||||||
| Function / homology |  Function and homology information Function and homology informationendoplasmic reticulum signal sequence receptor activity / SRP-dependent cotranslational protein targeting to membrane / signal recognition particle, endoplasmic reticulum targeting / protein targeting to ER / 7S RNA binding / SRP-dependent cotranslational protein targeting to membrane / Neutrophil degranulation / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.53 Å X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.53 Å | ||||||
 Authors Authors | Birse, D. / Kapp, U. / Strub, K. / Cusack, S. / Aberg, A. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 1997 Title: The crystal structure of the signal recognition particle Alu RNA binding heterodimer, SRP9/14. Authors: Birse, D.E. / Kapp, U. / Strub, K. / Cusack, S. / Aberg, A. #1: Journal: FEBS Lett. / Year: 1996Title: Crystallization and Preliminary Crystallographic Analysis of the Signal Recognition Particle Srpphi14-9 Fusion Protein Authors: Birse, D.E. / Doublie, S. / Kapp, U. / Strub, K. / Cusack, S. / Aberg, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
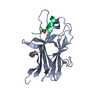
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1914.cif.gz | 49.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1914.ent.gz | 34.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1914.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/91/1914ftp://data.pdbj.org/pub/pdb/validation_reports/91/1914 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|







-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 26389.631 Da / Num. of mol.: 1 / Fragment: ALU RNA BINDING HETERODIMER Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #3: Chemical | ChemComp-BME /   Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 17 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 17 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 10 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 34 % Description: DATA WERE COLLECTED ON SELENIUM, MERCURY AND PLATINUM DERIVATIVES TO SOLVE MIR STRUCTURE | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: hanging drop / pH: 7.7 Details: THE SRPPHI14-9 PROTEIN WAS CRYSTALLIZED (BIRSE ET AL., FEBS LETTERS, 1996) BY THE HANGING DROP METHOD IN 2.0 M NAH2/K2H PO4, PH 7.7, 2% MPD, 1.0 MM NAN3 AT 4 DEGREES C WITH A FINAL PROTEIN ...Details: THE SRPPHI14-9 PROTEIN WAS CRYSTALLIZED (BIRSE ET AL., FEBS LETTERS, 1996) BY THE HANGING DROP METHOD IN 2.0 M NAH2/K2H PO4, PH 7.7, 2% MPD, 1.0 MM NAN3 AT 4 DEGREES C WITH A FINAL PROTEIN CONCENTRATION OF 5-8 MG ML-1. CRYSTALS FORMED OVER 2-3 WEEKS AND WERE TYPICALLY 150 X 150 X 300 MM3 IN SPACE, hanging drop, temperature 277K | ||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID2 / Wavelength: 0.9 / Wavelength: 0.900, 1.100 / Beamline: ID2 / Wavelength: 0.9 / Wavelength: 0.900, 1.100 | |||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1996 / Details: TOROIDAL MIRRORS | |||||||||
| Radiation | Monochromator: CU FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.53→30 Å / Num. obs: 36596 / % possible obs: 98.7 % / Observed criterion σ(I): 3 / Redundancy: 5 % / Rmerge(I) obs: 0.069 / Rsym value: 0.069 / Net I/σ(I): 5.8 | |||||||||
| Reflection shell | Resolution: 2.53→2.6 Å / Redundancy: 4.2 % / Rmerge(I) obs: 0.0218 / Mean I/σ(I) obs: 2.4 / Rsym value: 0.0218 / % possible all: 97.2 | |||||||||
| Reflection | *PLUS Num. obs: 7634 / Num. measured all: 36956 | |||||||||
| Reflection shell | *PLUS Lowest resolution: 2.64 Å / % possible obs: 97.2 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 2.53→20 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Cross valid method: THROUGHOUT / σ(F): 2 Details: THE MODEL INCLUDES 77 RESIDUES OF SRP9 (4-81) AND 84 RESIDUES OF SRP14 (1-34) AND (47-97). THE SRP14 LOOP ELECTRON DENSITY CONNECTING 14B1-14B2 (34-47) IS WEAK SUGGESTING THAT THE LOOP IS ...Details: THE MODEL INCLUDES 77 RESIDUES OF SRP9 (4-81) AND 84 RESIDUES OF SRP14 (1-34) AND (47-97). THE SRP14 LOOP ELECTRON DENSITY CONNECTING 14B1-14B2 (34-47) IS WEAK SUGGESTING THAT THE LOOP IS FLEXIBLE. 5 C-TERMINAL AND 3 N-TERMINAL RESIDUES OF SRP9 AND 13 C-TERMINAL RESIDUES OF SRP14 HAVE POORLY DEFINED DENSITY. IN ADDITION, THE MODEL DOES NOT INCLUDE THE SRPF14-9 N-TERMINAL F EXTENSION (18 RESIDUES) NOR THE ENTIRE LINKER REGION OF WHICH ONLY 8 OF 17 RESIDUES ARE ORDERED.
| |||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 20 Å | |||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.53→20 Å
| |||||||||||||||||||||
| LS refinement shell | Resolution: 2.53→2.64 Å / % reflection obs: 97.2 % | |||||||||||||||||||||
| Xplor file |
|
