Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3980: Post-catalytic P complex spliceosome with 3' splice site undocked -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3980 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Post-catalytic P complex spliceosome with 3' splice site undocked | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
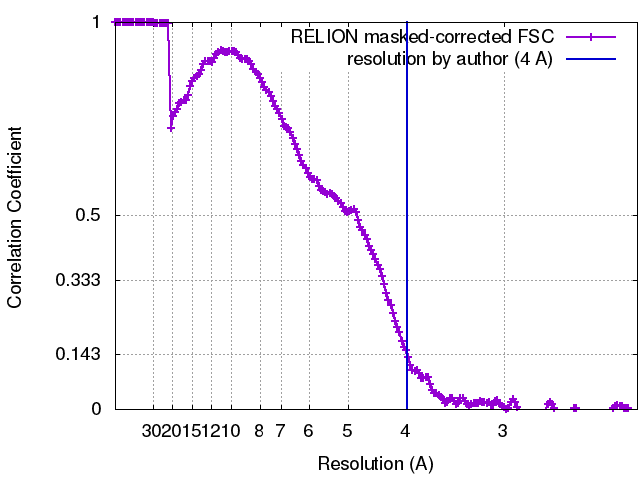
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | |||||||||
 Authors Authors | Wilkinson ME / Fica SM / Galej WP / Norman CM / Newman AJ / Nagai K | |||||||||
 Citation Citation | Journal: Science / Year: 2017 Title: Postcatalytic spliceosome structure reveals mechanism of 3'-splice site selection. Authors: Max E Wilkinson / Sebastian M Fica / Wojciech P Galej / Christine M Norman / Andrew J Newman / Kiyoshi Nagai /  Abstract: Introns are removed from eukaryotic messenger RNA precursors by the spliceosome in two transesterification reactions-branching and exon ligation. The mechanism of 3'-splice site recognition during ...Introns are removed from eukaryotic messenger RNA precursors by the spliceosome in two transesterification reactions-branching and exon ligation. The mechanism of 3'-splice site recognition during exon ligation has remained unclear. Here we present the 3.7-angstrom cryo-electron microscopy structure of the yeast P-complex spliceosome immediately after exon ligation. The 3'-splice site AG dinucleotide is recognized through non-Watson-Crick pairing with the 5' splice site and the branch-point adenosine. After the branching reaction, protein factors work together to remodel the spliceosome and stabilize a conformation competent for 3'-splice site docking, thereby promoting exon ligation. The structure accounts for the strict conservation of the GU and AG dinucleotides at the 5' and 3' ends of introns and provides insight into the catalytic mechanism of exon ligation. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3980.map.gz | 392.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3980-v30.xmlemd-3980.xml | 16.9 KB 16.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3980_fsc.xml | 17 KB | Display | FSC data file |
| Images |  emd_3980.png emd_3980.png | 68.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3980ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3980 http://ftp.pdbj.org/pub/emdb/structures/EMD-3980ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3980 | HTTPS FTP |
-Related structure data










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3980.map.gz / Format: CCP4 / Size: 421.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.12 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : P complex spliceosome with 3' splice site undocked
| Entire | Name: P complex spliceosome with 3' splice site undocked |
|---|---|
| Components |
|
-Supramolecule #1: P complex spliceosome with 3' splice site undocked
| Supramolecule | Name: P complex spliceosome with 3' splice site undocked / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#30 Details: P complex was assembled in vitro in splicing extract supplemented with dominant negative mutant Prp22 protein. P complex was purified via MS2-MBP fusion protein bound to 3 MS2 hairpin loops on the mRNA. |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 2.2 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.4 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.9 Component:
| ||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 7.0 nm / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK III Details: 3 microlitres sample were applied to the grid, left for 15 seconds and then blotted for 3.0 seconds before plunging.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 1-20 / Number grids imaged: 1 / Number real images: 2295 / Average exposure time: 12.0 sec. / Average electron dose: 47.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 0.2 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |