Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3502: Structural reorganization of the chromatin remodeling enzyme Chd1... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3502 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes | |||||||||
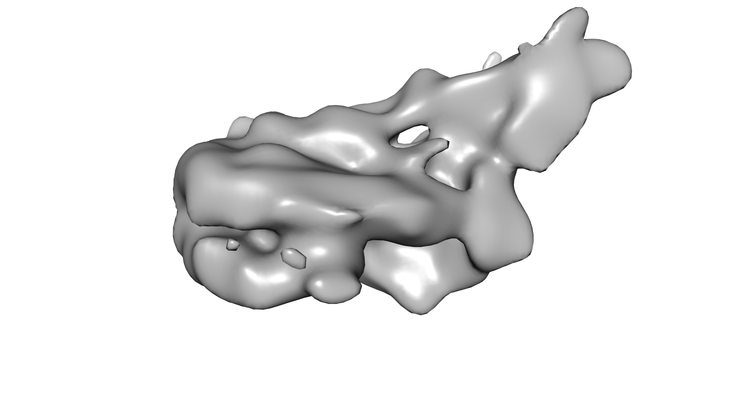
 Map data Map data | Structure of S.cerevisiae chromatin remodeling enzyme Chd1 bound to Nucleosome. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
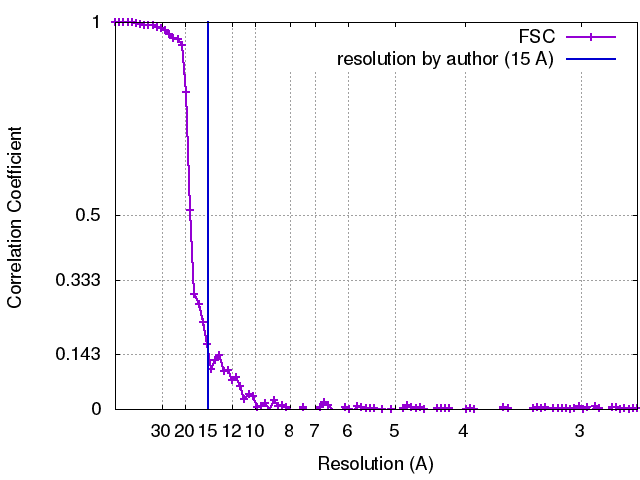
| Method | single particle reconstruction / cryo EM / Resolution: 15.0 Å | |||||||||
 Authors Authors | Sundaramoorthy R / Owen-Hughes T | |||||||||
 Citation Citation | Journal: Elife / Year: 2017 Title: Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. Authors: Ramasubramanian Sundaramoorthy / Amanda L Hughes / Vijender Singh / Nicola Wiechens / Daniel P Ryan / Hassane El-Mkami / Maxim Petoukhov / Dmitri I Svergun / Barbara Treutlein / Salina Quack ...Authors: Ramasubramanian Sundaramoorthy / Amanda L Hughes / Vijender Singh / Nicola Wiechens / Daniel P Ryan / Hassane El-Mkami / Maxim Petoukhov / Dmitri I Svergun / Barbara Treutlein / Salina Quack / Monika Fischer / Jens Michaelis / Bettina Böttcher / David G Norman / Tom Owen-Hughes /   Abstract: The yeast Chd1 protein acts to position nucleosomes across genomes. Here, we model the structure of the Chd1 protein in solution and when bound to nucleosomes. In the apo state, the DNA-binding ...The yeast Chd1 protein acts to position nucleosomes across genomes. Here, we model the structure of the Chd1 protein in solution and when bound to nucleosomes. In the apo state, the DNA-binding domain contacts the edge of the nucleosome while in the presence of the non-hydrolyzable ATP analog, ADP-beryllium fluoride, we observe additional interactions between the ATPase domain and the adjacent DNA gyre 1.5 helical turns from the dyad axis of symmetry. Binding in this conformation involves unravelling the outer turn of nucleosomal DNA and requires substantial reorientation of the DNA-binding domain with respect to the ATPase domains. The orientation of the DNA-binding domain is mediated by sequences in the N-terminus and mutations to this part of the protein have positive and negative effects on Chd1 activity. These observations indicate that the unfavorable alignment of C-terminal DNA-binding region in solution contributes to an auto-inhibited state. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3502.map.gz | 55.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3502-v30.xmlemd-3502.xml | 21.8 KB 21.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3502_fsc.xml | 10.5 KB | Display | FSC data file |
| Images |  emd_3502.png emd_3502.png | 54 KB | ||
| Others | emd_3502_half_map_1.map.gzemd_3502_half_map_2.map.gz | 46.2 MB 46.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3502 http://ftp.pdbj.org/pub/emdb/structures/EMD-3502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3502 | HTTPS FTP |
-Related structure data















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_3502.map.gz / Format: CCP4 / Size: 59.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of S.cerevisiae chromatin remodeling enzyme Chd1 bound to Nucleosome. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.34 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: Half map1 of the reconstruction
| File | emd_3502_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map1 of the reconstruction | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Half map2 of the reconstruction
| File | emd_3502_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map2 of the reconstruction | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Chd1-Nucleosome complex
| Entire | Name: Chd1-Nucleosome complex |
|---|---|
| Components |
|
-Supramolecule #1: Chd1-Nucleosome complex
| Supramolecule | Name: Chd1-Nucleosome complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: S.cerevisiae chromatin remodelling enzyme in complex with Nucleosome |
|---|---|
| Molecular weight | Theoretical: 400 KDa |
-Supramolecule #2: chd1
| Supramolecule | Name: chd1 / type: complex / ID: 2 / Parent: 1 |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism:  |
-Supramolecule #3: nucleosome
| Supramolecule | Name: nucleosome / type: complex / ID: 3 / Parent: 1 |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
-Macromolecule #1: Chromodomain-Helicase-DNA containing protein
| Macromolecule | Name: Chromodomain-Helicase-DNA containing protein / type: other / ID: 1 / Classification: other |
|---|---|
| Sequence | String: MAAKDISTEV LQNPELYGLR RSHRAAAHQQ NYFNDSDDED DEDNIKQSRR KRMTTIEDDE DEFEDEEGE EDSGEDEDEE DFEEDDDYYG SPIKQNRSKP KSRTKSKSKS KPKSQSEKQS T VKIPTRFS NRQNKTVNYN IDYSDDDLLE SEDDYGSEEA LSEENVHEAS ...String: MAAKDISTEV LQNPELYGLR RSHRAAAHQQ NYFNDSDDED DEDNIKQSRR KRMTTIEDDE DEFEDEEGE EDSGEDEDEE DFEEDDDYYG SPIKQNRSKP KSRTKSKSKS KPKSQSEKQS T VKIPTRFS NRQNKTVNYN IDYSDDDLLE SEDDYGSEEA LSEENVHEAS ANPQPEDFHG ID IVINHRL KTSLEEGKVL EKTVPDLNNC KENYEFLIKW TDESHLHNTW ETYESIGQVR GLK RLDNYC KQFIIEDQQV RLDPYVTAED IEIMDMERER RLDEFEEFHV PERIIDSQRA SLED GTSQL QYLVKWRRLN YDEATWENAT DIVKLAPEQV KHFQNRENSK ILPQYSSNYT SQRPR FEKL SVQPPFIKGG ELRDFQLTGI NWMAFLWSKG DNGILADEMG LGKTVQTVAF ISWLIF ARR QNGPHIIVVP LSTMPAWLDT FEKWAPDLNC ICYMGNQKSR DTIREYEFYT NPRAKGK KT MKFNVLLTTY EYILKDRAEL GSIKWQFMAV DEAHRLKNAE SSLYESLNSF KVANRMLI T GTPLQNNIKE LAALVNFLMP GRFTIDQEID FENQDEEQEE YIHDLHRRIQ PFILRRLKK DVEKSLPSKT ERILRVELSD VQTEYYKNIL TKNYSALTAG AKGGHFSLLN IMNELKKASN HPYLFDNAE ERVLQKFGDG KMTRENVLRG LIMSSGKMVL LDQLLTRLKK DGHRVLIFSQ M VRMLDILG DYLSIKGINF QRLDGTVPSA QRRISIDHFN SPDSNDFVFL LSTRAGGLGI NL MTADTVV IFDSDWNPQA DLQAMARAHR IGQKNHVMVY RLVSKDTVEE EVLERARKKM ILE YAIISL GVTDGNKYTK KNEPNAGELS AILKFGAGNM FTATDNQKKL EDLNLDDVLN HAED HVTTP DLGESHLGGE EFLKQFEVTD YKADIDWDDI IPEEELKKLQ DEEQKRKDEE YVKEQ LEMM NRRDNALKKI KNSVNGDGTA ANSDSDDDST SRSSRRRARA NDMDSIGESE VRALYK AIL KFGNLKEILD ELIADGTLPV KSFEKYGETY DEMMEAAKDC VHEEEKNRKE ILEKLEK HA TAYRAKLKSG EIKAENQPKD NPLTRLSLKK REKKAVLFNF KGVKSLNAES LLSRVEDL K YLKNLINSNY KDDPLKFSLG NNTPKPVQNW SSNWTKEEDE KLLIGVFKYG YGSWTQIRD DPFLGITDKI FLNEVHNPVA KKSASSSDTT PTPSKKGKGI TGSSKKVPGA IHLGRRVDYL LSFLRGGLN TKSPSADIGS KKLPTGPSKK RQRKPANHSK SMTPEI |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: Solutions were made fresh | |||||||||
| Grid | Model: Quantifoil / Material: COPPER/RHODIUM / Mesh: 400 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 101.325 kPa | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100.00 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV | |||||||||
| Details | This sample was monodisperse. One Chd1 enzyme bound to the Nucleosome |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Digitization - Sampling interval: 14.0 µm / Digitization - Frames/image: 4-18 / Number grids imaged: 1 / Number real images: 2560 / Average exposure time: 1.0 sec. / Average electron dose: 2.27 e/Å2 Details: Images are collected in movie mode at 22 images per second |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 4.0 µm / Calibrated defocus min: 1.8 µm / Calibrated magnification: 59000 / Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 4.0 µm / Nominal defocus min: 1.8 µm / Nominal magnification: 59000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |