 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3407: Refinement of atomic models in high resolution EM reconstructions... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3407 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment | |||||||||
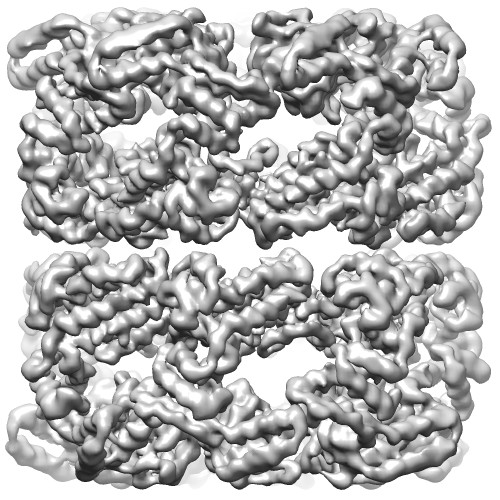
 Map data Map data | Unsharpened C7 reconstruction of GroEL | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Cryo-EM / Refinement of atomic models / assessment | |||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding ...GroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 3.26 Å | |||||||||
 Authors Authors | Joseph AP / Malhotra S / Burnley T / Wood C / Clare DK / Winn M / Topf M | |||||||||
 Citation Citation | Journal: Methods / Year: 2016 Title: Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. Authors: Agnel Praveen Joseph / Sony Malhotra / Tom Burnley / Chris Wood / Daniel K Clare / Martyn Winn / Maya Topf /  Abstract: As the resolutions of Three Dimensional Electron Microscopic reconstructions of biological macromolecules are being improved, there is a need for better fitting and refinement methods at high ...As the resolutions of Three Dimensional Electron Microscopic reconstructions of biological macromolecules are being improved, there is a need for better fitting and refinement methods at high resolutions and robust approaches for model assessment. Flex-EM/MODELLER has been used for flexible fitting of atomic models in intermediate-to-low resolution density maps of different biological systems. Here, we demonstrate the suitability of the method to successfully refine structures at higher resolutions (2.5-4.5Å) using both simulated and experimental data, including a newly processed map of Apo-GroEL. A hierarchical refinement protocol was adopted where the rigid body definitions are relaxed and atom displacement steps are reduced progressively at successive stages of refinement. For the assessment of local fit, we used the SMOC (segment-based Manders' overlap coefficient) score, while the model quality was checked using the Qmean score. Comparison of SMOC profiles at different stages of refinement helped in detecting regions that are poorly fitted. We also show how initial model errors can have significant impact on the goodness-of-fit. Finally, we discuss the implementation of Flex-EM in the CCP-EM software suite. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3407.map.gz | 82.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3407-v30.xmlemd-3407.xml | 10.5 KB 10.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3407_fsc.xml | 10.2 KB | Display | FSC data file |
| Images |  EMD-3407image.jpg EMD-3407image.jpg | 91 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3407ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3407 http://ftp.pdbj.org/pub/emdb/structures/EMD-3407ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3407 | HTTPS FTP |
-Related structure data











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_3407.map.gz / Format: CCP4 / Size: 100.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened C7 reconstruction of GroEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.055 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : GroEL
| Entire | Name: GroEL |
|---|---|
| Components |
|
-Supramolecule #1000: GroEL
| Supramolecule | Name: GroEL / type: sample / ID: 1000 / Oligomeric state: tetradecamer / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 56 KDa / Theoretical: 56 KDa |
-Macromolecule #1: GroEL
| Macromolecule | Name: GroEL / type: protein_or_peptide / ID: 1 / Name.synonym: 60 kDa Chaperonin / Oligomeric state: tetradecamer / Recombinant expression: No |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 800 KDa / Theoretical: 800 KDa |
| Sequence | UniProtKB: Chaperonin GroEL / GO: GroEL-GroES complex / InterPro: Chaperonin Cpn60/GroEL |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: 50 mM Tris-HCl, 50 mM KCl and 10 mM MgCl2 |
| Staining | Type: NEGATIVE Details: 3.5ul of protein was added to glow discharged C-flat r2/2 grid which were then blotted and plunge frozen |
| Grid | Details: 400 mesh C-flat r2/2 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 40 % / Chamber temperature: 100 K / Instrument: HOMEMADE PLUNGER Method: 3.5ul of protein was added to glow discharged C-flat r2/2 grid which were then blotted and plunge frozen |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 90 K / Max: 91 K / Average: 90 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at the magnification used to collect the data |
| Specialist optics | Energy filter - Name: Gatan / Energy filter - Lower energy threshold: 0.0 eV / Energy filter - Upper energy threshold: 20.0 eV |
| Details | The images were recorded using a dose rate of ~8 electrons/A2/s (~9 electrons/pixel/s) with 20 movie frames collected over a 4 second exposure |
| Date | Oct 9, 2015 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Digitization - Sampling interval: 5 µm / Number real images: 370 / Average electron dose: 32 e/Å2 Details: Each image is summation of 20 frames recorded over 4 seconds Bits/pixel: 32 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 47400 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.8 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | GroEL was aligned and reconstructed in Relion |
|---|---|
| CTF correction | Details: Each particle |
| Final reconstruction | Applied symmetry - Point group: C7 (7 fold cyclic) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 3.26 Å / Resolution method: OTHER / Software - Name: Relion, Ctffind3 / Number images used: 17400 |
| FSC plot (resolution estimation) | 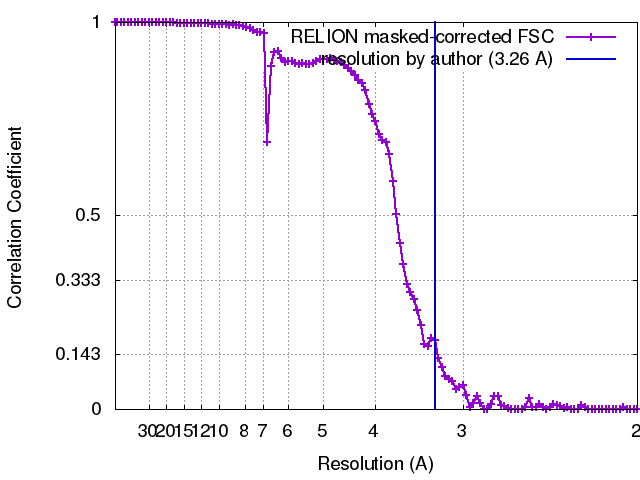 |
