 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-3407 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment | |||||||||
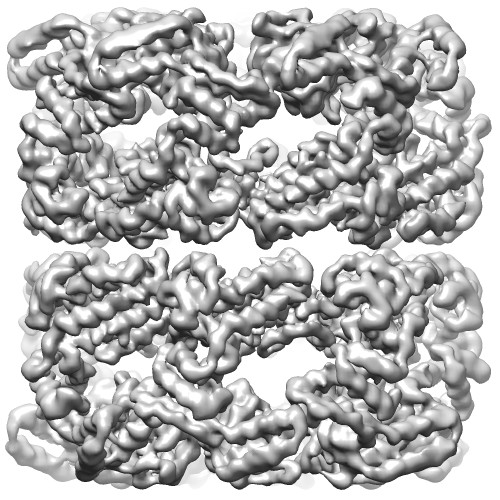
 マップデータ マップデータ | Unsharpened C7 reconstruction of GroEL | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード |  Cryo-EM (低温電子顕微鏡法) / Refinement of atomic models / assessment Cryo-EM (低温電子顕微鏡法) / Refinement of atomic models / assessment | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報GroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / フォールディング / response to heat ...GroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / フォールディング / response to heat / protein refolding / magnesium ion binding / ATP hydrolysis activity / ATP binding / 生体膜 / identical protein binding / 細胞質基質類似検索 - 分子機能 | |||||||||
| 生物種 |  Escherichia coli (大腸菌) Escherichia coli (大腸菌) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / ネガティブ染色法 / 解像度: 3.26 Å | |||||||||
 データ登録者 データ登録者 | Joseph AP / Malhotra S / Burnley T / Wood C / Clare DK / Winn M / Topf M | |||||||||
 引用 引用 | ジャーナル: Methods / 年: 2016 タイトル: Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. 著者: Agnel Praveen Joseph / Sony Malhotra / Tom Burnley / Chris Wood / Daniel K Clare / Martyn Winn / Maya Topf /  要旨: As the resolutions of Three Dimensional Electron Microscopic reconstructions of biological macromolecules are being improved, there is a need for better fitting and refinement methods at high ...As the resolutions of Three Dimensional Electron Microscopic reconstructions of biological macromolecules are being improved, there is a need for better fitting and refinement methods at high resolutions and robust approaches for model assessment. Flex-EM/MODELLER has been used for flexible fitting of atomic models in intermediate-to-low resolution density maps of different biological systems. Here, we demonstrate the suitability of the method to successfully refine structures at higher resolutions (2.5-4.5Å) using both simulated and experimental data, including a newly processed map of Apo-GroEL. A hierarchical refinement protocol was adopted where the rigid body definitions are relaxed and atom displacement steps are reduced progressively at successive stages of refinement. For the assessment of local fit, we used the SMOC (segment-based Manders' overlap coefficient) score, while the model quality was checked using the Qmean score. Comparison of SMOC profiles at different stages of refinement helped in detecting regions that are poorly fitted. We also show how initial model errors can have significant impact on the goodness-of-fit. Finally, we discuss the implementation of Flex-EM in the CCP-EM software suite. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_3407.map.gz | 82.6 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-3407-v30.xmlemd-3407.xml | 10.5 KB 10.5 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_3407_fsc.xml | 10.2 KB | 表示 | FSCデータファイル |
| 画像 |  EMD-3407image.jpg EMD-3407image.jpg | 91 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3407ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3407 http://ftp.pdbj.org/pub/emdb/structures/EMD-3407ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3407 | HTTPS FTP |
-関連構造データ











-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_3407.map.gz / 形式: CCP4 / 大きさ: 100.6 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened C7 reconstruction of GroEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.055 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
- 試料の構成要素
試料の構成要素
-全体 : GroEL
| 全体 | 名称: GroEL |
|---|---|
| 要素 |
|
-超分子 #1000: GroEL
| 超分子 | 名称: GroEL / タイプ: sample / ID: 1000 / 集合状態: tetradecamer / Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 56 KDa / 理論値: 56 KDa |
-分子 #1: GroEL
| 分子 | 名称: GroEL / タイプ: protein_or_peptide / ID: 1 / Name.synonym: 60 kDa Chaperonin / 集合状態: tetradecamer / 組換発現: No |
|---|---|
| 由来(天然) | 生物種: Escherichia coli (大腸菌) / 細胞中の位置: cytoplasm |
| 分子量 | 実験値: 800 KDa / 理論値: 800 KDa |
| 配列 | UniProtKB: Chaperonin GroEL / GO: GroEL-GroES complex / InterPro: Chaperonin Cpn60/GroEL |
-実験情報
-構造解析
| 手法 | ネガティブ染色法, クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 4 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 / 詳細: 50 mM Tris-HCl, 50 mM KCl and 10 mM MgCl2 |
| 染色 | タイプ: NEGATIVE 詳細: 3.5ul of protein was added to glow discharged C-flat r2/2 grid which were then blotted and plunge frozen |
| グリッド | 詳細: 400 mesh C-flat r2/2 |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 40 % / チャンバー内温度: 100 K / 装置: HOMEMADE PLUNGER 手法: 3.5ul of protein was added to glow discharged C-flat r2/2 grid which were then blotted and plunge frozen |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 47400 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / 最大 デフォーカス(公称値): 2.8 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 130000 |
| 特殊光学系 | エネルギーフィルター - 名称: Gatan エネルギーフィルター - エネルギー下限: 0.0 eV エネルギーフィルター - エネルギー上限: 20.0 eV |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 温度 | 最低: 90 K / 最高: 91 K / 平均: 90 K |
| アライメント法 | Legacy - 非点収差: Objective lens astigmatism was corrected at the magnification used to collect the data |
| 詳細 | The images were recorded using a dose rate of ~8 electrons/A2/s (~9 electrons/pixel/s) with 20 movie frames collected over a 4 second exposure |
| 日付 | 2015年10月9日 |
| 撮影 | カテゴリ: CCD フィルム・検出器のモデル: GATAN K2 QUANTUM (4k x 4k) デジタル化 - サンプリング間隔: 5 µm / 実像数: 370 / 平均電子線量: 32 e/Å2 詳細: Each image is summation of 20 frames recorded over 4 seconds ビット/ピクセル: 32 |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| CTF補正 | 詳細: Each particle |
|---|---|
| 最終 再構成 | 想定した対称性 - 点群: C7 (7回回転対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 3.26 Å / 解像度の算出法: OTHER / ソフトウェア - 名称: Relion, Ctffind3 / 使用した粒子像数: 17400 |
| 詳細 | GroEL was aligned and reconstructed in Relion |
| FSC曲線 (解像度の算出) | 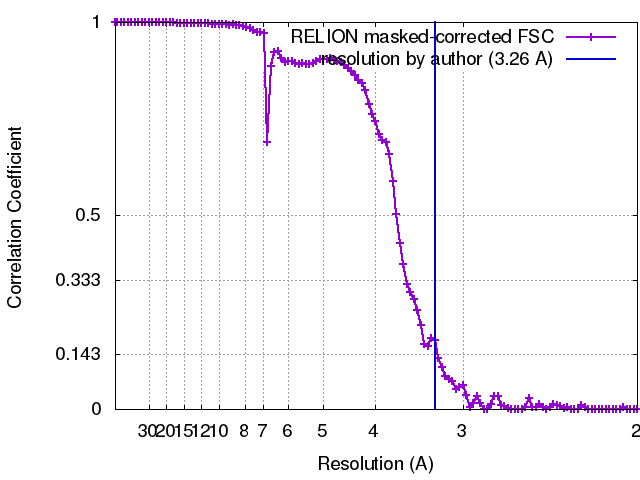 |
