 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-11600: Cubic core of the dihydrolipoamide acyltransferase (E2b) componen... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-11600 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cubic core of the dihydrolipoamide acyltransferase (E2b) component of the branched-chain alpha-ketoacid dehydrogenase complex (BCKDH) from M. tuberculosis | |||||||||
 Map data Map data | CryoEM sharpened map obtained from Crysoparc | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationdihydrolipoyllysine-residue (2-methylpropanoyl)transferase / dihydrolipoamide branched chain acyltransferase activity / peptidoglycan-based cell wall / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Mycobacterium tuberculosis H37Rv (bacteria) Mycobacterium tuberculosis H37Rv (bacteria) | |||||||||
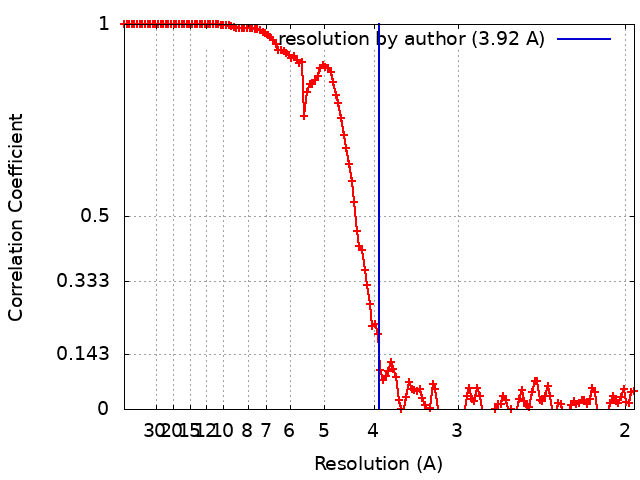
| Method | single particle reconstruction / cryo EM / Resolution: 3.92 Å | |||||||||
 Authors Authors | Bruch EM / Vilela P / Bellinzoni M | |||||||||
| Funding support |  France, 1 items France, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2021 Title: Actinobacteria challenge the paradigm: A unique protein architecture for a well-known, central metabolic complex. Authors: Eduardo M Bruch / Pierre Vilela / Lu Yang / Alexandra Boyko / Norik Lexa-Sapart / Bertrand Raynal / Pedro M Alzari / Marco Bellinzoni /  Abstract: α-oxoacid dehydrogenase complexes are large, tripartite enzymatic machineries carrying out key reactions in central metabolism. Extremely conserved across the tree of life, they have been, so far, ...α-oxoacid dehydrogenase complexes are large, tripartite enzymatic machineries carrying out key reactions in central metabolism. Extremely conserved across the tree of life, they have been, so far, all considered to be structured around a high-molecular weight hollow core, consisting of up to 60 subunits of the acyltransferase component. We provide here evidence that Actinobacteria break the rule by possessing an acetyltranferase component reduced to its minimally active, trimeric unit, characterized by a unique C-terminal helix bearing an actinobacterial specific insertion that precludes larger protein oligomerization. This particular feature, together with the presence of an gene coding for both the decarboxylase and the acyltransferase domains on the same polypetide, is spread over Actinobacteria and reflects the association of PDH and ODH into a single physical complex. Considering the central role of the pyruvate and 2-oxoglutarate nodes in central metabolism, our findings pave the way to both therapeutic and metabolic engineering applications. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_11600.map.gz | 223.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-11600-v30.xmlemd-11600.xml | 14.8 KB 14.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_11600_fsc.xml | 13.7 KB | Display | FSC data file |
| Images |  emd_11600.png emd_11600.png | 203.2 KB | ||
| Others | emd_11600_additional_1.map.gz | 10.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-11600ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11600 http://ftp.pdbj.org/pub/emdb/structures/EMD-11600ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11600 | HTTPS FTP |
-Related structure data
| Related structure data |  6zziC  6zzjC  6zzkC  6zzlC  6zzmC  6zznC C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_11600.map.gz / Format: CCP4 / Size: 236.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM sharpened map obtained from Crysoparc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.97 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
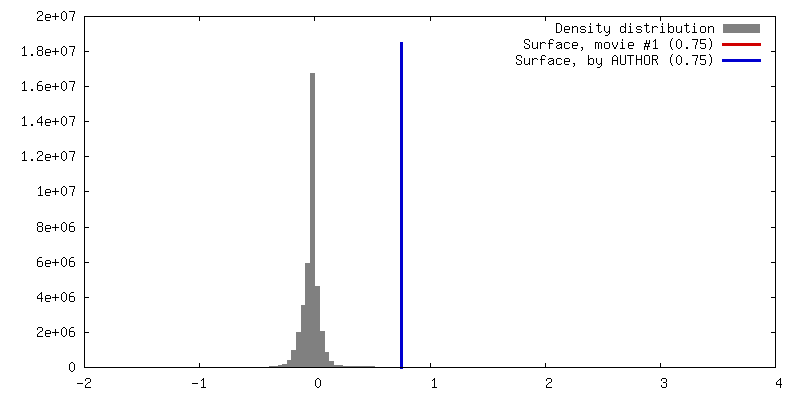
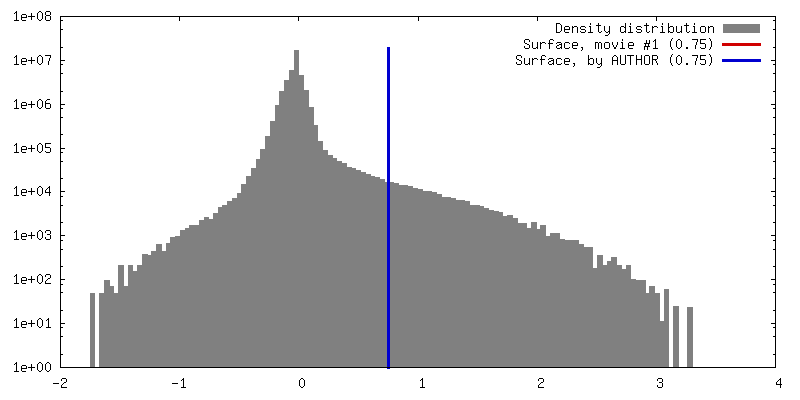
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Additional map: Map after density modification using ResolveEM in Phenix suite
| File | emd_11600_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map after density modification using ResolveEM in Phenix suite | ||||||||||||
| Projections & Slices |
| ||||||||||||
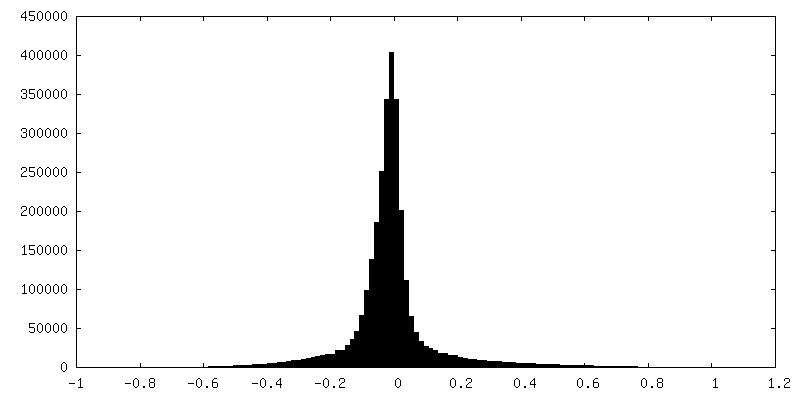
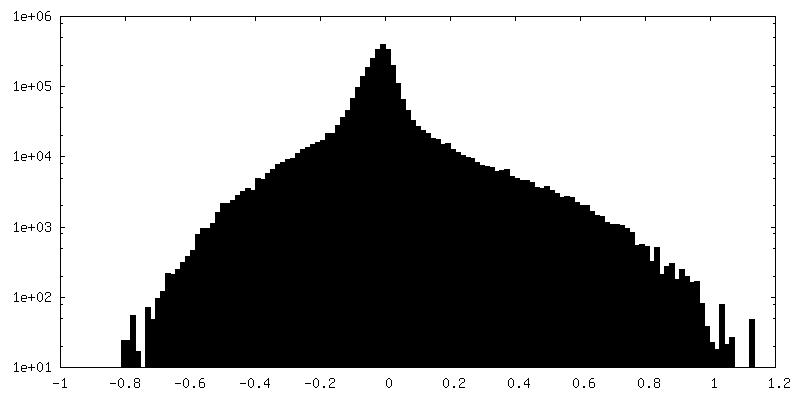
| Density Histograms |






- Sample components
Sample components
-Entire : Catalytic domain of M. tuberculosis E2b (acyltransferase from bra...
| Entire | Name: Catalytic domain of M. tuberculosis E2b (acyltransferase from branched-chain alpha-ketoacid dehydrogenase complex) |
|---|---|
| Components |
|
-Supramolecule #1: Catalytic domain of M. tuberculosis E2b (acyltransferase from bra...
| Supramolecule | Name: Catalytic domain of M. tuberculosis E2b (acyltransferase from branched-chain alpha-ketoacid dehydrogenase complex) type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Mycobacterium tuberculosis H37Rv (bacteria) |
| Recombinant expression | Organism: |
| Molecular weight | Theoretical: 592 KDa |
-Macromolecule #1: Dihydrolipoyllysine-residue acyltransferase
| Macromolecule | Name: Dihydrolipoyllysine-residue acyltransferase / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO EC number: dihydrolipoyllysine-residue (2-methylpropanoyl)transferase |
|---|---|
| Source (natural) | Organism: Mycobacterium tuberculosis H37Rv (bacteria) |
| Recombinant expression | Organism: |
| Sequence | String: GSPDVRPVHG VHARMAEK M TLSHKEIPTA KASVEVICAE LLRLRDRFVS AAPEITPFAL TLRLLVIALK HNVILNSTW VDSGEGPQVH VHRGVHLGFG AATERGLLVP VVTDAQDKNT RELASRVAEL ITGAREGTLT PAELRGSTF TVSNFGALGV DDGVPVINHP ...String: GSPDVRPVHG VHARMAEK M TLSHKEIPTA KASVEVICAE LLRLRDRFVS AAPEITPFAL TLRLLVIALK HNVILNSTW VDSGEGPQVH VHRGVHLGFG AATERGLLVP VVTDAQDKNT RELASRVAEL ITGAREGTLT PAELRGSTF TVSNFGALGV DDGVPVINHP EAAILGLGAI KPRPVVVGGE VVARPTMTLT C VFDHRVVD GAQVAQFMCE LRDLIESPET ALLDL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.6 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8.5 Component:
| |||||||||
| Grid | Material: COPPER / Mesh: 200 / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER / Details: Power 5W | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: Blot time 4 seconds before plunging. |
- Electron microscopy
Electron microscopy
| Microscope | TFS GLACIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: INTEGRATING / Number grids imaged: 2 / Number real images: 5009 / Average exposure time: 1.0 sec. / Average electron dose: 42.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 150000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 165-393 |
|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
