 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0908: Cryo EM map of b0,+ AT-rBAT complex bound with Arginine, focused ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0908 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo EM map of b0,+ AT-rBAT complex bound with Arginine, focused refined on TM domain | ||||||||||||||||||||||||
 Map data Map data | Cryo EM map of b0, AT-rBAT complex bound with Arginine, focused refined on TM domain | ||||||||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.6 Å | ||||||||||||||||||||||||
 Authors Authors | Yan RH / Li YN / Lei JL / Zhou Q | ||||||||||||||||||||||||
| Funding support |  China, 7 items China, 7 items
| ||||||||||||||||||||||||
 Citation Citation | Journal: Sci Adv / Year: 2020 Title: Cryo-EM structure of the human heteromeric amino acid transporter bAT-rBAT. Authors: Renhong Yan / Yaning Li / Yi Shi / Jiayao Zhou / Jianlin Lei / Jing Huang / Qiang Zhou / Abstract: Heteromeric amino acid transporters (HATs) catalyze the transmembrane movement of amino acids, comprising two subunits, a heavy chain and a light chain, linked by a disulfide bridge. The bAT (SLC7A9) ...Heteromeric amino acid transporters (HATs) catalyze the transmembrane movement of amino acids, comprising two subunits, a heavy chain and a light chain, linked by a disulfide bridge. The bAT (SLC7A9) is a representative light chain of HATs, forming heterodimer with rBAT, a heavy chain which mediates the membrane trafficking of bAT. The bAT-rBAT complex is an obligatory exchanger, which mediates the influx of cystine and cationic amino acids and the efflux of neutral amino acids in kidney and small intestine. Here, we report the cryo-EM structure of the human bAT-rBAT complex alone and in complex with arginine substrate at resolution of 2.7 and 2.3 Å, respectively. The overall structure of bAT-rBAT exists as a dimer of heterodimer consistent with the previous study. A ligand molecule is bound to the substrate binding pocket, near which an occluded pocket is identified, to which we found that it is important for substrate transport. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0908.map.gz | 116.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0908-v30.xmlemd-0908.xml | 12.5 KB 12.5 KB | Display Display | EMDB header |
| Images | 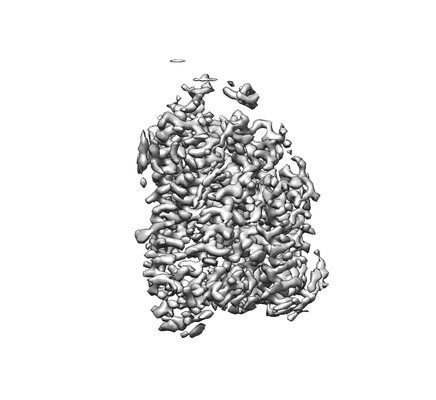 emd_0908.png emd_0908.png | 37.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0908ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0908 http://ftp.pdbj.org/pub/emdb/structures/EMD-0908ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0908 | HTTPS FTP |
-Related structure data
| Related structure data |  0903C  0904C  0905C  0906C  0907C  6li9C  6lidC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0908.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo EM map of b0, AT-rBAT complex bound with Arginine, focused refined on TM domain | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.091 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : b0,+AT-rBAT complex bound with Arginine, focused refined on TM domain
| Entire | Name: b0,+AT-rBAT complex bound with Arginine, focused refined on TM domain |
|---|---|
| Components |
|
-Supramolecule #1: b0,+AT-rBAT complex bound with Arginine, focused refined on TM domain
| Supramolecule | Name: b0,+AT-rBAT complex bound with Arginine, focused refined on TM domain type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 120 KDa |
-Macromolecule #1: rBAT
| Macromolecule | Name: rBAT / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MAHHHHHHHH HHSGRAEDKS KRDSIEMSMK GCQTNNGFVH NEDILEQTPD PGSSTDNLKH STRGILGSQE PDFKGVQPYA GMPKEVLFQF SGQARYRIPR EILFWLTVAS VLVLIAATIA IIALSPKCLD WWQEGPMYQI YPRSFKDSNK DGNGDLKGIQ DKLDYITALN ...String: MAHHHHHHHH HHSGRAEDKS KRDSIEMSMK GCQTNNGFVH NEDILEQTPD PGSSTDNLKH STRGILGSQE PDFKGVQPYA GMPKEVLFQF SGQARYRIPR EILFWLTVAS VLVLIAATIA IIALSPKCLD WWQEGPMYQI YPRSFKDSNK DGNGDLKGIQ DKLDYITALN IKTVWITSFY KSSLKDFRYG VEDFREVDPI FGTMEDFENL VAAIHDKGLK LIIDFIPNHT SDKHIWFQLS RTRTGKYTDY YIWHDCTHEN GKTIPPNNWL SVYGNSSWHF DEVRNQCYFH QFMKEQPDLN FRNPDVQEEI KEILRFWLTK GVDGFSLDAV KFLLEAKHLR DEIQVNKTQI PDTVTQYSEL YHDFTTTQVG MHDIVRSFRQ TMDQYSTEPG RYRFMGTEAY AESIDRTVMY YGLPFIQEAD FPFNNYLSML DTVSGNSVYE VITSWMENMP EGKWPNWMIG GPDSSRLTSR LGNQYVNVMN MLLFTLPGTP ITYYGEEIGM GNIVAANLNE SYDINTLRSK SPMQWDNSSN AGFSEASNTW LPTNSDYHTV NVDVQKTQPR SALKLYQDLS LLHANELLLN RGWFCHLRND SHYVVYTREL DGIDRIFIVV LNFGESTLLN LHNMISGLPA KMRIRLSTNS ADKGSKVDTS GIFLDKGEGL IFEHNTKNLL HRQTAFRDRC FVSNRACYSS VLNILYTSC |
-Macromolecule #2: b0,+AT
| Macromolecule | Name: b0,+AT / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MADYKDDDDK SGPDEVDASG RGDTGLRKRR EDEKSIQSQE PKTTSLQKEL GLISGISIIV GTIIGSGIFV SPKSVLSNTE AVGPCLIIWA ACGVLATLGA LCFAELGTMI TKSGGEYPYL MEAYGPIPAY LFSWASLIVI KPTSFAIICL SFSEYVCAPF YVGCKPPQIV ...String: MADYKDDDDK SGPDEVDASG RGDTGLRKRR EDEKSIQSQE PKTTSLQKEL GLISGISIIV GTIIGSGIFV SPKSVLSNTE AVGPCLIIWA ACGVLATLGA LCFAELGTMI TKSGGEYPYL MEAYGPIPAY LFSWASLIVI KPTSFAIICL SFSEYVCAPF YVGCKPPQIV VKCLAAAAIL FISTVNSLSV RLGSYVQNIF TAAKLVIVAI IIISGLVLLA QGNTKNFDNS FEGAQLSVGA ISLAFYNGLW AYDGWNQLNY ITEELRNPYR NLPLAIIIGI PLVTACYILM NVSYFTVMTA TELLQSQAVA VTFGDRVLYP ASWIVPLFVA FSTIGAANGT CFTAGRLIYV AGREGHMLKV LSYISVRRLT PAPAIIFYGI IATIYIIPGD INSLVNYFSF AAWLFYGLTI LGLIVMRFTR KELERPIKVP VVIPVLMTLI SVFLVLAPII SKPTWEYLYC VLFILSGLLF YFLFVHYKFG WAQKISKPIT MHLQMLMEVV PPEEDPE |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 |
|---|---|
| Vitrification | Cryogen name: NITROGEN |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 2.6 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3) / Number images used: 1331654 |
|---|---|
| Initial angle assignment | Type: OTHER |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
