 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3337 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
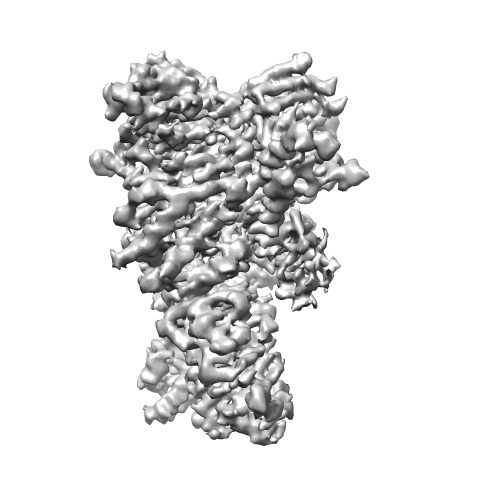
| Title | Atomic cryoEM structure of Hsp90/Cdc37/Cdk4 complex | |||||||||
 Map data Map data | Reconstruction of Hsp90:Cdc37:Cdk4. Part of series of maps, the highest resolution one out of the series. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Hsp90 / Cdc37 / Cdk4 / chaperone / kinase / unfolding | |||||||||
| Function / homology |  Function and homology information Function and homology informationcyclin D3-CDK4 complex / cyclin D1-CDK4 complex / cyclin D2-CDK4 complex / Evasion of Oncogene Induced Senescence Due to Defective p16INK4A binding to CDK4 / Evasion of Oxidative Stress Induced Senescence Due to Defective p16INK4A binding to CDK4 / citrulline metabolic process / regulation of transcription initiation by RNA polymerase II / regulation of type II interferon-mediated signaling pathway / Evasion of Oncogene Induced Senescence Due to Defective p16INK4A binding to CDK4 and CDK6 / Evasion of Oxidative Stress Induced Senescence Due to Defective p16INK4A binding to CDK4 and CDK6 ...cyclin D3-CDK4 complex / cyclin D1-CDK4 complex / cyclin D2-CDK4 complex / Evasion of Oncogene Induced Senescence Due to Defective p16INK4A binding to CDK4 / Evasion of Oxidative Stress Induced Senescence Due to Defective p16INK4A binding to CDK4 / citrulline metabolic process / regulation of transcription initiation by RNA polymerase II / regulation of type II interferon-mediated signaling pathway / Evasion of Oncogene Induced Senescence Due to Defective p16INK4A binding to CDK4 and CDK6 / Evasion of Oxidative Stress Induced Senescence Due to Defective p16INK4A binding to CDK4 and CDK6 / Drug-mediated inhibition of CDK4/CDK6 activity / regulation of type B pancreatic cell proliferation / HSP90-CDC37 chaperone complex / very long-chain fatty acid metabolic process / negative regulation of proteasomal protein catabolic process / Aryl hydrocarbon receptor signalling / aryl hydrocarbon receptor complex / cellular response to ionomycin / dynein axonemal particle / histone methyltransferase binding / Transcriptional regulation by RUNX2 / : / cellular response to phorbol 13-acetate 12-myristate / receptor ligand inhibitor activity / positive regulation of type 2 mitophagy / ATP-dependent protein binding / positive regulation of protein localization to cell surface / regulation of cyclin-dependent protein serine/threonine kinase activity / protein kinase regulator activity / protein folding chaperone complex / cyclin-dependent protein serine/threonine kinase regulator activity / post-transcriptional regulation of gene expression / Respiratory syncytial virus genome replication / telomerase holoenzyme complex assembly / positive regulation of transforming growth factor beta receptor signaling pathway / Uptake and function of diphtheria toxin / PTK6 Regulates Cell Cycle / Drug-mediated inhibition of ERBB2 signaling / Resistance of ERBB2 KD mutants to trastuzumab / Resistance of ERBB2 KD mutants to sapitinib / Resistance of ERBB2 KD mutants to tesevatinib / Resistance of ERBB2 KD mutants to neratinib / Resistance of ERBB2 KD mutants to osimertinib / Resistance of ERBB2 KD mutants to afatinib / Resistance of ERBB2 KD mutants to AEE788 / Resistance of ERBB2 KD mutants to lapatinib / Drug resistance in ERBB2 TMD/JMD mutants / regulation of type I interferon-mediated signaling pathway / TPR domain binding / dendritic growth cone / Defective binding of RB1 mutants to E2F1,(E2F2, E2F3) / Assembly and release of respiratory syncytial virus (RSV) virions / The NLRP3 inflammasome / protein phosphatase activator activity / Sema3A PAK dependent Axon repulsion / regulation of protein ubiquitination / HSF1-dependent transactivation / response to unfolded protein / bicellular tight junction / cyclin-dependent kinase / HSF1 activation / cyclin-dependent protein serine/threonine kinase activity / axonal growth cone / telomere maintenance via telomerase / protein targeting / Attenuation phase / chaperone-mediated protein complex assembly / RHOBTB2 GTPase cycle / Purinergic signaling in leishmaniasis infection / cyclin-dependent protein kinase holoenzyme complex / supramolecular fiber organization / : / regulation of G2/M transition of mitotic cell cycle / heat shock protein binding / DNA polymerase binding / positive regulation of G2/M transition of mitotic cell cycle / Signaling by ERBB2 / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / peptide binding / protein folding chaperone / cellular response to interleukin-4 / cyclin binding / nitric-oxide synthase regulator activity / placenta development / ESR-mediated signaling / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / Constitutive Signaling by Overexpressed ERBB2 / Ubiquitin-dependent degradation of Cyclin D / positive regulation of cell differentiation / Hsp90 protein binding / ATP-dependent protein folding chaperone / Signaling by ERBB2 TMD/JMD mutants / G1/S transition of mitotic cell cycle / Constitutive Signaling by EGFRvIII / Signaling by ERBB2 ECD mutants / DDX58/IFIH1-mediated induction of interferon-alpha/beta / Oncogene Induced Senescence / Signaling by ERBB2 KD Mutants / Meiotic recombination / Regulation of actin dynamics for phagocytic cup formation Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.9 Å | |||||||||
 Authors Authors | Verba KA / Wang RYR / Arakawa A / Liu Y / Shirouzu M / Yokoyama S / Agard DA | |||||||||
 Citation Citation | Journal: Science / Year: 2016 Title: Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Authors: Kliment A Verba / Ray Yu-Ruei Wang / Akihiko Arakawa / Yanxin Liu / Mikako Shirouzu / Shigeyuki Yokoyama / David A Agard /   Abstract: The Hsp90 molecular chaperone and its Cdc37 cochaperone help stabilize and activate more than half of the human kinome. However, both the mechanism by which these chaperones assist their "client" ...The Hsp90 molecular chaperone and its Cdc37 cochaperone help stabilize and activate more than half of the human kinome. However, both the mechanism by which these chaperones assist their "client" kinases and the reason why some kinases are addicted to Hsp90 while closely related family members are independent are unknown. Our structural understanding of these interactions is lacking, as no full-length structures of human Hsp90, Cdc37, or either of these proteins with a kinase have been elucidated. Here we report a 3.9 angstrom cryo-electron microscopy structure of the Hsp90-Cdc37-Cdk4 kinase complex. Surprisingly, the two lobes of Cdk4 are completely separated with the β4-β5 sheet unfolded. Cdc37 mimics part of the kinase N lobe, stabilizing an open kinase conformation by wedging itself between the two lobes. Finally, Hsp90 clamps around the unfolded kinase β5 strand and interacts with exposed N- and C-lobe interfaces, protecting the kinase in a trapped unfolded state. On the basis of this structure and an extensive amount of previously collected data, we propose unifying conceptual and mechanistic models of chaperone-kinase interactions. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3337.map.gz | 59.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3337-v30.xmlemd-3337.xml | 16.9 KB 16.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3337_fsc.xml | 9 KB | Display | FSC data file |
| Images | emd_3337.tif | 118.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3337ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3337 http://ftp.pdbj.org/pub/emdb/structures/EMD-3337ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3337 | HTTPS FTP |
-Validation report
| Summary document | emd_3337_validation.pdf.gz | 305.1 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_3337_full_validation.pdf.gz | 304.2 KB | Display | |
| Data in XML | emd_3337_validation.xml.gz | 10.4 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3337ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3337 | HTTPS FTP |
-Related structure data
| Related structure data |  5fwkMC  3338C  3339C  3340C  3341C  3342C  3343C  3344C  5fwlC  5fwmC  5fwpC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |
























-Map
| File | Download / File: emd_3337.map.gz / Format: CCP4 / Size: 62.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of Hsp90:Cdc37:Cdk4. Part of series of maps, the highest resolution one out of the series. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.315 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Complex of Human Hsp90 beta, human Cdc37 and human Cdk4
| Entire | Name: Complex of Human Hsp90 beta, human Cdc37 and human Cdk4 |
|---|---|
| Components |
|
-Supramolecule #1000: Complex of Human Hsp90 beta, human Cdc37 and human Cdk4
| Supramolecule | Name: Complex of Human Hsp90 beta, human Cdc37 and human Cdk4 type: sample / ID: 1000 / Details: All three proteins were co-expressed in Sf9 cells. Oligomeric state: One Hsp90 homodimer binds to one Cdc37 and one Cdk4 Number unique components: 3 |
|---|---|
| Molecular weight | Experimental: 245 KDa / Theoretical: 245 KDa / Method: As cloned, verified by SDS-PAGE |
-Macromolecule #1: Heat Shock Protein HSP 90 beta
| Macromolecule | Name: Heat Shock Protein HSP 90 beta / type: protein_or_peptide / ID: 1 / Name.synonym: Hsp90 / Number of copies: 2 / Oligomeric state: Dimer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / synonym: Human / Location in cell: cytoplasm |
| Molecular weight | Theoretical: 83 KDa |
| Recombinant expression | Organism:   Spodoptera frugiperda (fall armyworm) / Recombinant plasmid: pFastBacHT Spodoptera frugiperda (fall armyworm) / Recombinant plasmid: pFastBacHT |
| Sequence | UniProtKB: Heat shock protein HSP 90-beta / GO: citrulline metabolic process / InterPro: Heat shock protein Hsp90 family |
-Macromolecule #2: Hsp90 co-chaperone Cdc37
| Macromolecule | Name: Hsp90 co-chaperone Cdc37 / type: protein_or_peptide / ID: 2 / Name.synonym: Cdc37 / Number of copies: 1 / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / synonym: Human / Location in cell: throughout |
| Molecular weight | Theoretical: 44.5 KDa |
| Recombinant expression | Organism: Spodoptera frugiperda (fall armyworm) / Recombinant plasmid: pFastBacHT |
| Sequence | UniProtKB: Hsp90 co-chaperone Cdc37 / GO: GO: 0000002 |
-Macromolecule #3: Cyclin-dependent kinase 4
| Macromolecule | Name: Cyclin-dependent kinase 4 / type: protein_or_peptide / ID: 3 / Name.synonym: Cdk4 / Number of copies: 1 / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / synonym: Human / Location in cell: throughout |
| Molecular weight | Theoretical: 33.7 KDa |
| Recombinant expression | Organism: Spodoptera frugiperda (fall armyworm) / Recombinant plasmid: pFastBacHT |
| Sequence | UniProtKB: Cyclin-dependent kinase 4 / GO: very long-chain fatty acid metabolic process / InterPro: Protein kinase domain |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.27 mg/mL |
|---|---|
| Buffer | pH: 7.5 Details: 20mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM KCl, 10 mM MgCl2, 20 mM Na2MoO4, 2mM DTT, 0.085mM DDM |
| Grid | Details: Glow discharged for 30 sec, C-flat 400 mesh 1.2/1.3 thick carbon grids (Protochips) |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 95 K / Instrument: FEI VITROBOT MARK III / Method: Single blot from 4 to 6 seconds, at 20C |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Alignment procedure | Legacy - Astigmatism: At high mag via FT. |
| Date | Nov 25, 2014 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Number real images: 3718 / Average electron dose: 44 e/Å2 / Details: 38 frames, 7.6 seconds total exposure / Bits/pixel: 8 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.8 µm / Nominal defocus min: 1.4 µm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | Image stacks were corrected for motion and summed as described previously, resulting in binned sums (1.315A/pix). For particle picking the images were binned to 5.2A/pix and Gaussian bandpass filtered between 15A and 500A using EMAN2. SamViewer template based picking was then used to pick particles from all the micrographs, followed by manual review of all the picks. After such procedure 802877 particles were picked in total and extracted from images binned to 2.6A/pix. CTFFIND4 was used to estimate defocus parameters for all the images. Relion 1.4 was used for all the following steps unless noted otherwise. Reference free 2D classification into 300 classes for 75 iterations was performed followed by manual examination of the resulting class averages. Low resolution/signal to noise/feature class averages and contributing particles were discarded, resulting in 670000 particles left. The resulting particles were 3D classified into 4 classes resulting in two classes having high-resolution features (390000 particles). At this stage particles were extracted from 1.315A/pix micrographs and all the following processing was done with these particles. Using 3D Auto-refine in Relion 1.4, a reconstruction was obtained from 390000 particles resulting from 3D classification above (using highest resolution 3D class as initial model, low pass filtered to 20A). Using the resulting parameters, the particles were further drift corrected per particle and dose weighted using the Particle Polishing feature. The B-factor weighing curve was fit by a polynomial (with a rationale that such a curve should be smooth) and used to generate new weighting parameters for Particle Polishing, with which 390000 particles were then polished. All further data processing was done using the polished particles. Re-refinement of the 390000 particles after polishing yielded the map at about 4A resolution (determined using gold standard FSC in the PostProcessing tab). Raw particles were sharpened with a B-factor of -50, low pass filtered with Gaussian filter to 3A and the refinement was continued for 10 more iterations (until convergence) with these particles (the rationale was that due to extremely low noise levels of K2 direct detector, this would yield more accurate alignments due to presence of more high resolution data in the images). This resulted in similar resolution but one with visually sharper features. Lastly, this reconstruction was tightly masked around Hsp90 region and refinement was further continued for 4 iterations (until convergence), with rationale that Hsp90 region is more coherent. This yielded a 3.9A reconstruction with the best density for Hsp90 core region. |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.9 Å / Resolution method: OTHER / Software - Name: Relion / Number images used: 388688 |
| Final two d classification | Number classes: 1 |
| FSC plot (resolution estimation) | 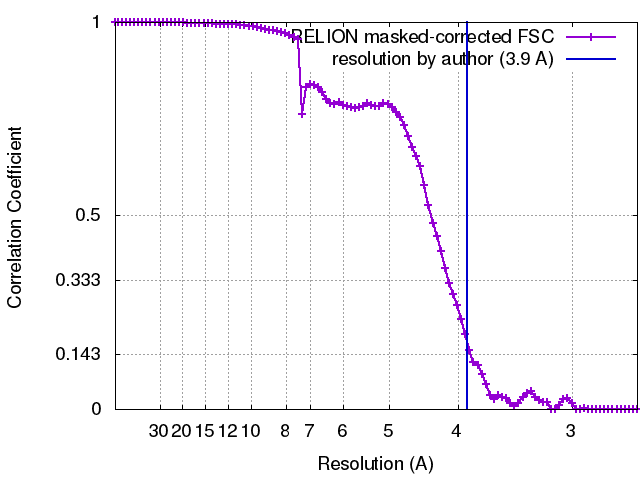 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B |
|---|---|
| Software | Name: Rosetta, MDFF, Chimera, COLORES (Situs) |
| Details | Atomic model building and refinement the Hsp90/Cdc37/Cdk4 complex was performed incrementally in five stages: 1) de novo model-building for Cdc37, 2) structure refinement of the Hsp90/Cdc37 complex, 3) de novo model extension for Cdk4 in the presence of the refined Hsp90/Cdc37 complex, and 4) structure refinement of the Hsp90/Cdc37/Cdk4 complex. The atomic structure of Hsp90/Cdc37/Cdk4 complex was used in the modelling of other low-resolution maps. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 95 Target criteria: compound of Rosetta energy function and electron density fitting function |
| Output model | PDB-5fwk: |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: Rosetta, MDFF, Chimera, COLORES (Situs) |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 95 Target criteria: compound of Rosetta energy function and electron density fitting function |
| Output model | PDB-5fwk: |

