National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01-GM084192
United States
Citation
Journal: Nat Struct Mol Biol / Year: 2022 Title: Nucleosome recognition and DNA distortion by the Chd1 remodeler in a nucleotide-free state. Authors: Ilana M Nodelman / Sayan Das / Anneliese M Faustino / Stephen D Fried / Gregory D Bowman / Jean-Paul Armache / Abstract: Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free ...Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free state, determined by cryo-EM to 2.3 Å resolution. The remodeler stimulates the nucleosome to absorb an additional nucleotide on each strand at two different locations: on the tracking strand within the ATPase binding site and on the guide strand one helical turn from the ATPase motor. Remarkably, the additional nucleotide on the tracking strand is associated with a local transformation toward an A-form geometry, explaining how sequential ratcheting of each DNA strand occurs. The structure also reveals a histone-binding motif, ChEx, which can block opposing remodelers on the nucleosome and may allow Chd1 to participate in histone reorganization during transcription.
History
Deposition
Nov 20, 2021
-
Header (metadata) release
Mar 2, 2022
-
Map release
Mar 2, 2022
-
Update
Jan 17, 2024
-
Current status
Jan 17, 2024
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
EMPIAR-10876 (Title: 2.3 A structure of the ATP-dependent chromatin remodeler Chd1 bound to the nucleosome in a nucleotide-free state Data size: 4.7 TB Data #1: Unaligned frames of Chd1-bound nucleosomes collected on Gatan K3 in Super Resolution [micrographs - multiframe] Data #2: MotionCor2-aligned frames of Chd1-bound nucleosomes collected on Gatan K3 [micrographs - single frame] Data #3: Processed subsets [picked particles - single frame - processed])
Entire : Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
Entire
Name: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state
Components
Complex: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state
-
Supramolecule #1: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
Supramolecule
Name: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state type: complex / ID: 1 / Parent: 0 / Details: This entry has a weak DNA-binding domain
pH: 7 Details: 20 mM HEPES, pH 7.0, 60 mM KCl, 1.5 mM DTT, 1 mM MgCl2
Grid
Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 10 sec.
Vitrification
Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV
Details
Nucleosome-bound CHD1, prepared in the presence of ATPgammaS, and then crosslinked using GraFix.
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Specialist optics
Energy filter - Slit width: 20 eV
Image recording
Film or detector model: GATAN K3 (6k x 4k) / Digitization - Dimensions - Width: 11520 pixel / Digitization - Dimensions - Height: 8184 pixel / Number grids imaged: 1 / Number real images: 6906 / Average exposure time: 3.3 sec. / Average electron dose: 49.9 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links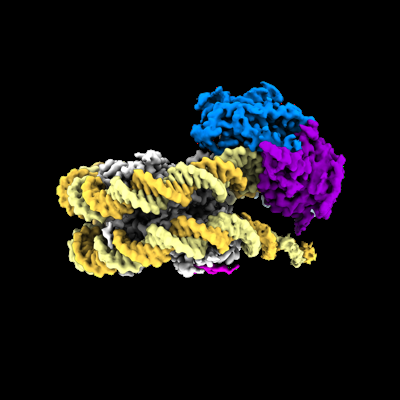 emd_25479.png
emd_25479.png http://ftp.pdbj.org/pub/emdb/structures/EMD-25479
http://ftp.pdbj.org/pub/emdb/structures/EMD-25479















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





































































 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN



