 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8179 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Campylobacter Hook | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationbacterial-type flagellum organization / bacterial-type flagellum basal body / bacterial-type flagellum-dependent swarming motility Similarity search - Function | |||||||||
| Biological species |  Campylobacter jejuni subsp. jejuni 81116 (Campylobacter) Campylobacter jejuni subsp. jejuni 81116 (Campylobacter) | |||||||||
| Method | helical reconstruction / cryo EM / Resolution: 3.5 Å | |||||||||
 Authors Authors | Matsunami H / Wolf M / Samatey F | |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2016 Title: Complete structure of the bacterial flagellar hook reveals extensive set of stabilizing interactions. Authors: Hideyuki Matsunami / Clive S Barker / Young-Ho Yoon / Matthias Wolf / Fadel A Samatey /  Abstract: The bacterial flagellar hook is a tubular helical structure made by the polymerization of multiple copies of a protein, FlgE. Here we report the structure of the hook from Campylobacter jejuni by ...The bacterial flagellar hook is a tubular helical structure made by the polymerization of multiple copies of a protein, FlgE. Here we report the structure of the hook from Campylobacter jejuni by cryo-electron microscopy at a resolution of 3.5 Å. On the basis of this structure, we show that the hook is stabilized by intricate inter-molecular interactions between FlgE molecules. Extra domains in FlgE, found only in Campylobacter and in related bacteria, bring more stability and robustness to the hook. Functional experiments suggest that Campylobacter requires an unusually strong hook to swim without its flagella being torn off. This structure reveals details of the quaternary organization of the hook that consists of 11 protofilaments. Previous study of the flagellar filament of Campylobacter by electron microscopy showed its quaternary structure made of seven protofilaments. Therefore, this study puts in evidence the difference between the quaternary structures of a bacterial filament and its hook. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8179.map.gz | 56.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8179-v30.xmlemd-8179.xml | 15 KB 15 KB | Display Display | EMDB header |
| Images |  emd_8179.png emd_8179.png | 223.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8179ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8179 http://ftp.pdbj.org/pub/emdb/structures/EMD-8179ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8179 | HTTPS FTP |
-Related structure data
| Related structure data |  5jxlMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_8179.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
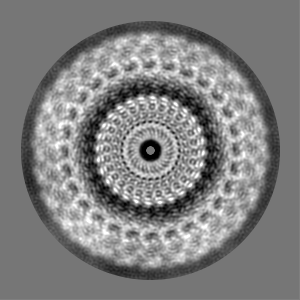
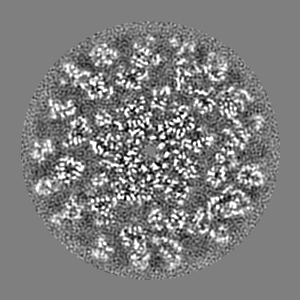
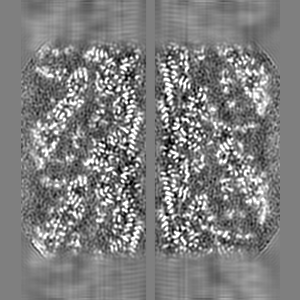
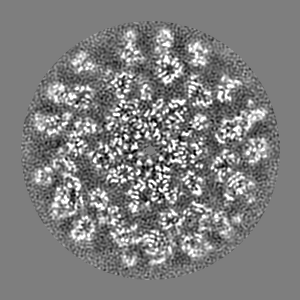
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
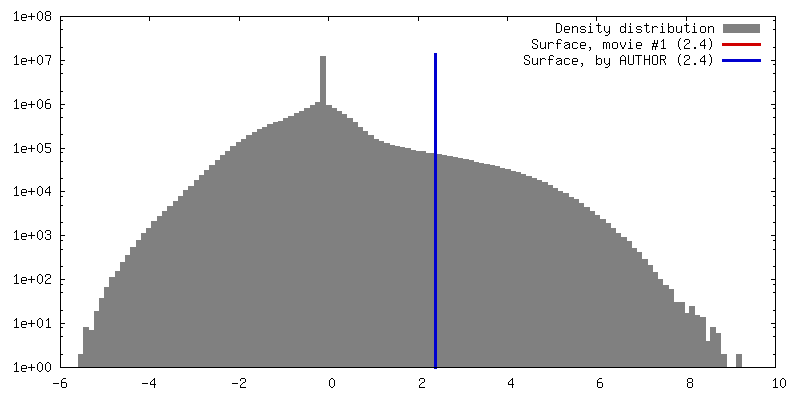
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















-Supplemental data
- Sample components
Sample components
-Entire : helical assembly of FlgE (flagellar hook)
| Entire | Name: helical assembly of FlgE (flagellar hook) |
|---|---|
| Components |
|
-Supramolecule #1: helical assembly of FlgE (flagellar hook)
| Supramolecule | Name: helical assembly of FlgE (flagellar hook) / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Campylobacter jejuni subsp. jejuni 81116 (Campylobacter) |
-Macromolecule #1: Campylobacter hook FlgE
| Macromolecule | Name: Campylobacter hook FlgE / type: protein_or_peptide / ID: 1 Details: cju:C8J_1629 hypothetical protein; K02390 flagellar hook protein FlgE (A) Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Campylobacter jejuni subsp. jejuni 81116 (Campylobacter) |
| Sequence | String: MRSLWSGVSG LQAHQVAMDV EGNNISNVNT TGFKYSRADF GTMFSQTVKI ATAPTDGRG GSNPLQIGLG VSVSSTTRIH SQGSVQTTDK NTDVAINGDG FFMVSDDGGL TNYLTRSGDF KLDAYGNFV NNAGFVVQGW NINWDDQTID SSRTPQNIFI DPGMHIPAAK ...String: MRSLWSGVSG LQAHQVAMDV EGNNISNVNT TGFKYSRADF GTMFSQTVKI ATAPTDGRG GSNPLQIGLG VSVSSTTRIH SQGSVQTTDK NTDVAINGDG FFMVSDDGGL TNYLTRSGDF KLDAYGNFV NNAGFVVQGW NINWDDQTID SSRTPQNIFI DPGMHIPAAK STEVAIKANL N SGLNIGTS SRNLYALDSV HGWNTKTQRA EDENDTGTTQ FYTTSKNSVE VTEKGVDAGA LF NANGTGL NLRDGQGIWV SYADAKFTTD RANGANVFDP NLTVAQQNNV IFWGNKDIAV TLD INLNGV RIQNDNIRSL DEAIAYINTF TAPTDTRDGT GVKAVKKADG SGIEFVNNNA DGTT DNMKN IDLTVNVGNS AGERNTINYN ANTGVFSPQG GNLTTAQNDT DWIAGAAQAG QPQNV KVVT AHKYIYSSNP VTIPPMINPD GGPAFQPNNG NRPTDPASAN YWDAIQGSLK NTTERT FRT TEDLRELLQR DARYGVDYNG SGIIDNATPT FDANDINQAV KVVVTENGNF AISNANE TS TIPANAGAGA GAATTNPKNM SFNITAYSNK QGTVSTNDAF TKIFKAFDGP LVIGNQIK E SEQLKLSAFS AGLEIYDSLG SKHTLEVQFV KQSTTQDGGN EWQMIIRVPE PAEINTTGE GPTNIIVGTA RFNNDGSLAN YTPKTINFSP NNGAAPNQQI KLSFGTSGSN DGLVSSNSAS TLTGQATDG YTSGNLKPDA IRVDDKGNIL GEFTNGKTFA VAKIAMASVA NNSGLEEIGG N LFKVTANS GNIVVGEAGT GGRGEMKTSA LEMSNVDLSR SLTELIIIQR GYQANSKTIS TS DQMLQTL IQLKQ |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | filament |
-Sample preparation
| Concentration | 0.2 mg/mL |
|---|---|
| Buffer | pH: 8 / Component - Concentration: 10.0 mM / Component - Formula: Tris-HCl / Component - Name: Tris Hydrochloride / Details: EDTA, Triton X-100 |
| Grid | Model: Protochips C-Flat CF-1.2/1.3 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Support film - Film thickness: 30.0 nm / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER / Pretreatment - Pressure: 1e-05 kPa |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 280 K / Instrument: GATAN CRYOPLUNGE 3 Details: blotting from both sides, Whatman #40 filter paper, 25 seconds blot time. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 63.0 K / Max: 70.0 K |
| Details | parallel illumination |
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Digitization - Dimensions - Width: 4000 pixel / Digitization - Dimensions - Height: 4000 pixel / Digitization - Sampling interval: 14.0 µm / Digitization - Frames/image: 1-5 / Number grids imaged: 1 / Number real images: 504 / Average exposure time: 1.5 sec. / Average electron dose: 2.4 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 2.5 µm / Calibrated defocus min: 0.5 µm / Calibrated magnification: 129629 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |

