 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-7065: Cryo-EM structure of human insulin degrading enzyme in complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7065 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of human insulin degrading enzyme in complex with insulin | |||||||||||||||
 Map data Map data | Insulin degrading enzyme in complex with insulin | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | IDE / insulin degrading enzyme / amyloid beta / BIOSYNTHETIC PROTEIN / HYDROLASE | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationinsulysin / beta-endorphin binding / ubiquitin recycling / insulin catabolic process / insulin metabolic process / amyloid-beta clearance by cellular catabolic process / hormone catabolic process / bradykinin catabolic process / cytosolic proteasome complex / positive regulation of protein binding ...insulysin / beta-endorphin binding / ubiquitin recycling / insulin catabolic process / insulin metabolic process / amyloid-beta clearance by cellular catabolic process / hormone catabolic process / bradykinin catabolic process / cytosolic proteasome complex / positive regulation of protein binding / insulin binding / regulation of aerobic respiration / peptide catabolic process / peroxisomal matrix / amyloid-beta clearance / amyloid-beta metabolic process / Insulin receptor recycling / negative regulation of proteolysis / peptide binding / protein catabolic process / : / Peroxisomal protein import / antigen processing and presentation of endogenous peptide antigen via MHC class I / metalloendopeptidase activity / positive regulation of protein catabolic process / insulin receptor signaling pathway / peroxisome / amyloid-beta binding / virus receptor activity / endopeptidase activity / basolateral plasma membrane / Ub-specific processing proteases / external side of plasma membrane / protein-containing complex binding / cell surface / protein homodimerization activity / ATP hydrolysis activity / mitochondrion / proteolysis / : / extracellular exosome / zinc ion binding / ATP binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||||||||
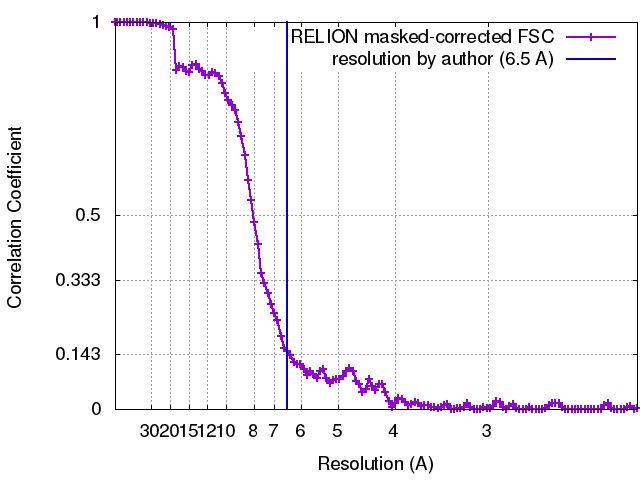
| Method | single particle reconstruction / cryo EM / Resolution: 6.5 Å | |||||||||||||||
 Authors Authors | Zhang Z / Liang WG / Bailey LJ / Tan YZ / Wei H / Kossiakoff AA / Carragher B / Potter SC / Tang WJ | |||||||||||||||
| Funding support |  United States, 4 items United States, 4 items
| |||||||||||||||
 Citation Citation | Journal: Elife / Year: 2018 Title: Ensemble cryoEM elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme. Authors: Zhening Zhang / Wenguang G Liang / Lucas J Bailey / Yong Zi Tan / Hui Wei / Andrew Wang / Mara Farcasanu / Virgil A Woods / Lauren A McCord / David Lee / Weifeng Shang / Rebecca Deprez- ...Authors: Zhening Zhang / Wenguang G Liang / Lucas J Bailey / Yong Zi Tan / Hui Wei / Andrew Wang / Mara Farcasanu / Virgil A Woods / Lauren A McCord / David Lee / Weifeng Shang / Rebecca Deprez-Poulain / Benoit Deprez / David R Liu / Akiko Koide / Shohei Koide / Anthony A Kossiakoff / Sheng Li / Bridget Carragher / Clinton S Potter / Wei-Jen Tang /  Abstract: Insulin degrading enzyme (IDE) plays key roles in degrading peptides vital in type two diabetes, Alzheimer's, inflammation, and other human diseases. However, the process through which IDE recognizes ...Insulin degrading enzyme (IDE) plays key roles in degrading peptides vital in type two diabetes, Alzheimer's, inflammation, and other human diseases. However, the process through which IDE recognizes peptides that tend to form amyloid fibrils remained unsolved. We used cryoEM to understand both the apo- and insulin-bound dimeric IDE states, revealing that IDE displays a large opening between the homologous ~55 kDa N- and C-terminal halves to allow selective substrate capture based on size and charge complementarity. We also used cryoEM, X-ray crystallography, SAXS, and HDX-MS to elucidate the molecular basis of how amyloidogenic peptides stabilize the disordered IDE catalytic cleft, thereby inducing selective degradation by substrate-assisted catalysis. Furthermore, our insulin-bound IDE structures explain how IDE processively degrades insulin by stochastically cutting either chain without breaking disulfide bonds. Together, our studies provide a mechanism for how IDE selectively degrades amyloidogenic peptides and offers structural insights for developing IDE-based therapies. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
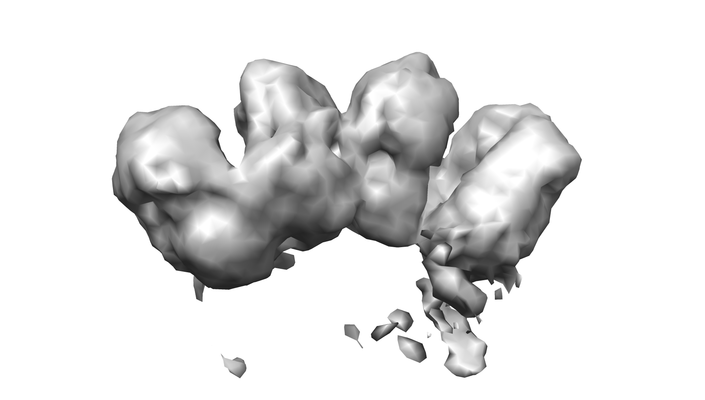
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7065.map.gz | 8.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7065-v30.xmlemd-7065.xml | 23.3 KB 23.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_7065_fsc.xml | 11.2 KB | Display | FSC data file |
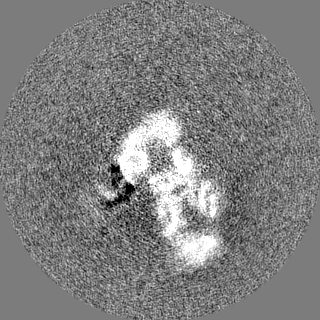
| Images |  emd_7065.png emd_7065.png | 66.1 KB | ||
| Masks | emd_7065_msk_1.map | 125 MB | Mask map | |
| Filedesc metadata | emd-7065.cif.gz | 6.9 KB | ||
| Others | emd_7065_half_map_1.map.gzemd_7065_half_map_2.map.gz | 98.3 MB 98.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7065ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7065 http://ftp.pdbj.org/pub/emdb/structures/EMD-7065ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7065 | HTTPS FTP |
-Related structure data
| Related structure data |  6b7yMC  7041C  7062C  7066C  7090C  7091C  7092C  7093C  5wobC  6b3qC  6b70C  6b7zC  6bf6C  6bf7C  6bf8C  6bf9C  6bfcC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_7065.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Insulin degrading enzyme in complex with insulin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.073 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_7065_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
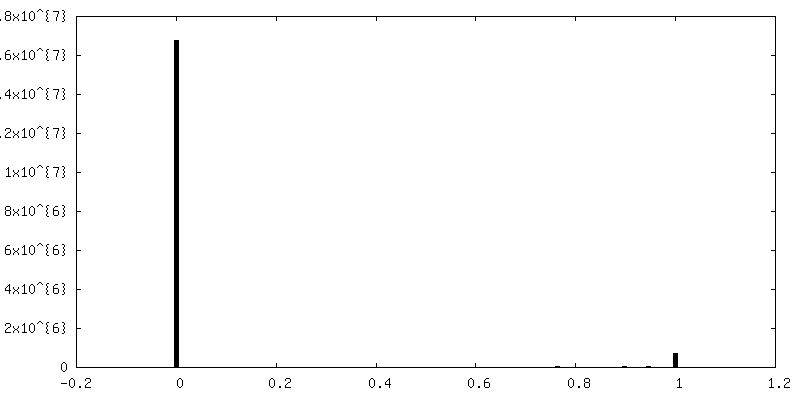
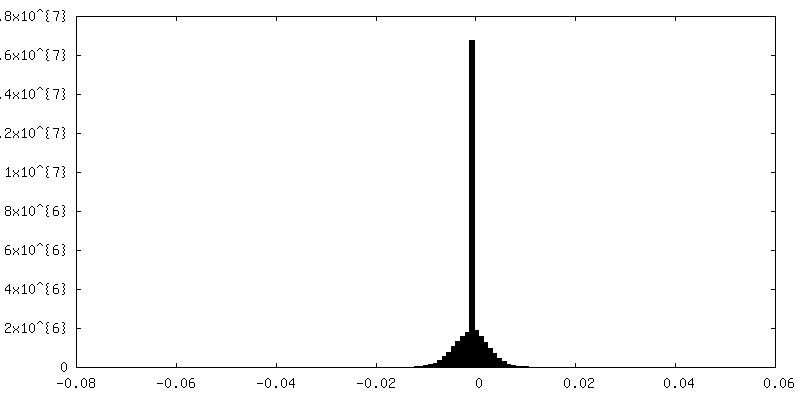
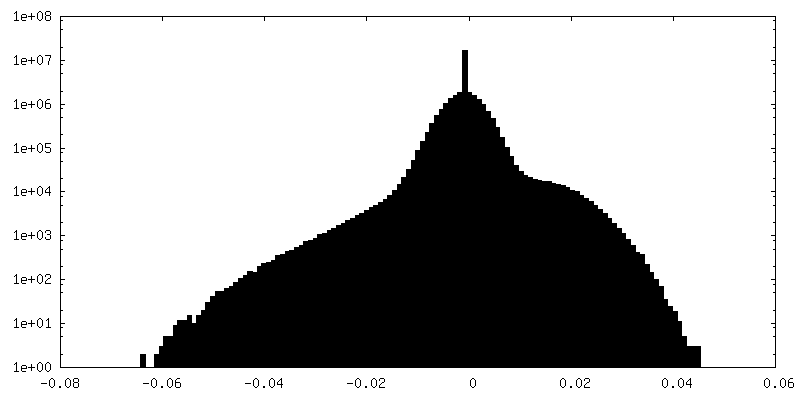
| Density Histograms |







-Half map: Insulin degrading enzyme in complex with insulin
| File | emd_7065_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Insulin degrading enzyme in complex with insulin | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
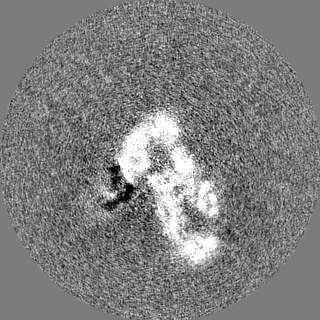
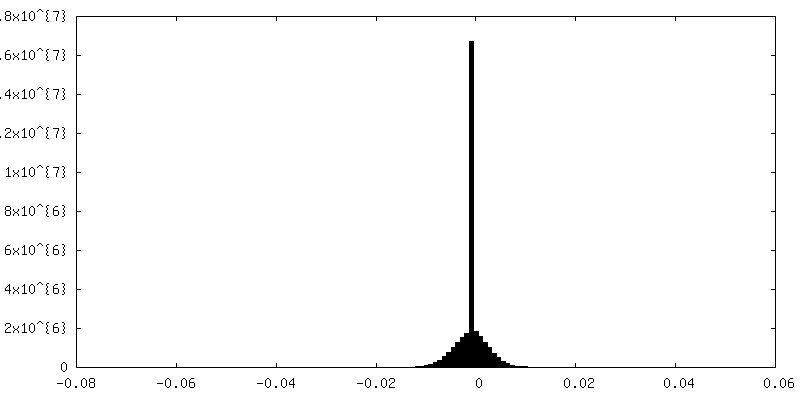
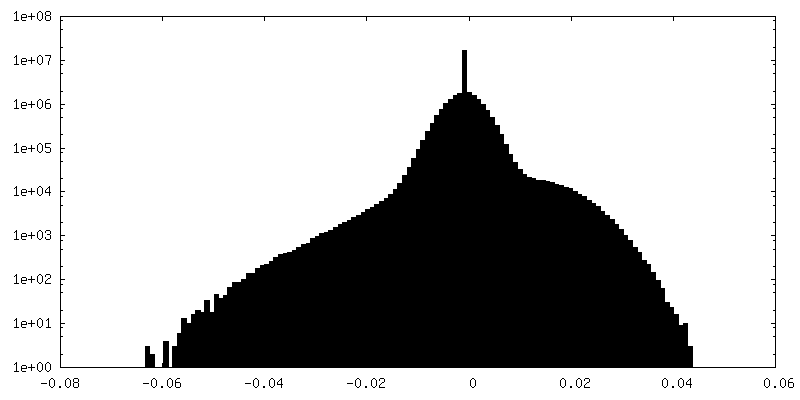
-Half map: Insulin degrading enzyme in complex with insulin
| File | emd_7065_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Insulin degrading enzyme in complex with insulin | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
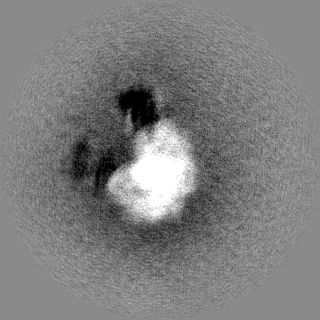
- Sample components
Sample components
-Entire : Insulin degrading enzyme
| Entire | Name: Insulin degrading enzyme |
|---|---|
| Components |
|
-Supramolecule #1: Insulin degrading enzyme
| Supramolecule | Name: Insulin degrading enzyme / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: Cryo-EM structure of human Apo insulin degrading enzyme |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 100 KDa |
-Macromolecule #1: Insulin-degrading enzyme
| Macromolecule | Name: Insulin-degrading enzyme / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO / EC number: insulysin |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 111.866484 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: AIKRIGNHIT KSPEDKREYR GLELANGIKV LLISDPTTDK SSAALDVHIG SLSDPPNIAG LSHFLEHMLF LGTKKYPKEN EYSQFLSEH AGSSNAFTSG EHTNYYFDVS HEHLEGALDR FAQFFLSPLF DESAKDREVN AVDSEHEKNV MNDAWRLFQL E KATGNPKH ...String: AIKRIGNHIT KSPEDKREYR GLELANGIKV LLISDPTTDK SSAALDVHIG SLSDPPNIAG LSHFLEHMLF LGTKKYPKEN EYSQFLSEH AGSSNAFTSG EHTNYYFDVS HEHLEGALDR FAQFFLSPLF DESAKDREVN AVDSEHEKNV MNDAWRLFQL E KATGNPKH PFSKFGTGNK YTLETRPNQE GIDVRQELLK FHSAYYSSNL MAVVVLGRES LDDLTNLVVK LFSEVENKNV PL PEFPEHP FQEEHLKQLY KIVPIKDIRN LYVTFPIPDL QKYYKSNPGH YLGHLIGHEG PGSLLSELKS KGWVNTLVGG QKE GARGFM FFIINVDLTE EGLLHVEDII LHMFQYIQKL RAEGPQEWVF QELKDLNAVA FRFKDKERPR GYTSKIAGIL HYYP LEEVL TAEYLLEEFR PDLIEMVLDK LRPENVRVAI VSKSFEGKTD RTEEWYGTQY KQEAIPDEVI KKWQNADLNG KFKLP TKNE FIPTNFEILP LEKEATPYPA LIKDTAMSKL WFKQDDKFFL PKANLNFEFF SPFAYVDPLH SNMAYLYLEL LKDSLN EYA YAAELAGLSY DLQNTIYGMY LSVKGYNDKQ PILLKKIIEK MATFEIDEKR FEIIKEAYMR SLNNFRAEQP HQHAMYY LR LLMTEVAWTK DELKEALDDV TLPRLKAFIP QLLSRLHIEA LLHGNITKQA ALGIMQMVED TLIEHAHTKP LLPSQLVR Y REVQLPDRGW FVYQQRNEVH NNSGIEIYYQ TDMQSTSENM FLELFAQIIS EPAFNTLRTK EQLGYIVFSG PRRANGIQG LRFIIQSEKP PHYLESRVEA FLITMEKSIE DMTEEAFQKH IQALAIRRLD KPKKLSAESA KYWGEIISQQ YNFDRDNTEV AYLKTLTKE DIIKFYKEML AVDAPRRHKV SVHVLAREMD SCPVVGEFPC QNDINLSQAP ALPQPEVIQN MTEFKRGLPL F PLVKPH UniProtKB: Insulin-degrading enzyme |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.3 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.8 Component:
| ||||||||||||
| Grid | Model: Homemade / Material: GOLD / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Support film - Film thickness: 10 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 10 sec. / Pretreatment - Atmosphere: OTHER / Pretreatment - Pressure: 0.001 kPa / Details: The grids are homemade lacey gold nanowire grids | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 85 % / Chamber temperature: 298 K / Instrument: HOMEMADE PLUNGER / Details: The cryo grids were made using Spotiton. | ||||||||||||
| Details | The sample was monodisperse |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Alignment procedure | Coma free - Residual tilt: 10.0 mrad |
| Details | The image was collected at 20-50 degree tilt |
| Image recording | #0 - Image recording ID: 1 / #0 - Film or detector model: GATAN K2 SUMMIT (4k x 4k) / #0 - Detector mode: COUNTING / #0 - Number grids imaged: 1 / #0 - Number real images: 509 / #0 - Average exposure time: 10.0 sec. / #0 - Average electron dose: 7.9 e/Å2 / #1 - Image recording ID: 2 / #1 - Film or detector model: GATAN K2 SUMMIT (4k x 4k) / #1 - Detector mode: COUNTING / #1 - Digitization - Dimensions - Width: 3710 pixel / #1 - Digitization - Dimensions - Height: 3838 pixel / #1 - Digitization - Frames/image: 1-50 / #1 - Number grids imaged: 1 / #1 - Number real images: 620 / #1 - Average exposure time: 10.0 sec. / #1 - Average electron dose: 6.8 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated magnification: 46598 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.2 µm / Nominal defocus min: 0.9400000000000001 µm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 92 |
|---|---|
| Output model | PDB-6b7y: |
