 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6wfs: Cryo-EM Structure of Hepatitis B virus T=4 capsid in complex with... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6wfs | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cryo-EM Structure of Hepatitis B virus T=4 capsid in complex with the antiviral molecule DBT1 | ||||||
 Components Components | Capsid protein | ||||||
 Keywords Keywords | VIRUS LIKE PARTICLE / capsid / antiviral / HBV | ||||||
| Function / homology |  Function and homology information Function and homology informationmicrotubule-dependent intracellular transport of viral material towards nucleus / T=4 icosahedral viral capsid / viral penetration into host nucleus / host cell / host cell cytoplasm / symbiont entry into host cell / structural molecule activity / DNA binding / RNA binding / identical protein binding Similarity search - Function | ||||||
| Biological species |  Hepatitis B virus genotype D subtype adw Hepatitis B virus genotype D subtype adw | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 4.6 Å | ||||||
 Authors Authors | Schlicksup, C. / Laughlin, P. / Dunkelbarger, S. / Wang, J.C. / Zlotnick, A. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: ACS Chem Biol / Year: 2020 Title: Local Stabilization of Subunit-Subunit Contacts Causes Global Destabilization of Hepatitis B Virus Capsids. Authors: Christopher John Schlicksup / Patrick Laughlin / Steven Dunkelbarger / Joseph Che-Yen Wang / Adam Zlotnick / Abstract: Development of antiviral molecules that bind virion is a strategy that remains in its infancy, and the details of their mechanisms are poorly understood. Here we investigate the behavior of DBT1, a ...Development of antiviral molecules that bind virion is a strategy that remains in its infancy, and the details of their mechanisms are poorly understood. Here we investigate the behavior of DBT1, a dibenzothiazepine that specifically interacts with the capsid protein of hepatitis B virus (HBV). We found that DBT1 stabilizes protein-protein interaction, accelerates capsid assembly, and can induce formation of aberrant particles. Paradoxically, DBT1 can cause preformed capsids to dissociate. These activities may lead to (i) assembly of empty and defective capsids, inhibiting formation of new virus, and (ii) disruption of mature viruses, which are metastable, to inhibit new infection. Using cryo-electron microscopy, we observed that DBT1 led to asymmetric capsids where well-defined DBT1 density was bound at all intersubunit contacts. These results suggest that DBT1 can support assembly by increasing buried surface area but induce disassembly of metastable capsids by favoring asymmetry to induce structural defects. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6wfs.cif.gz | 110.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6wfs.ent.gz | 88.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6wfs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wf/6wfsftp://data.pdbj.org/pub/pdb/validation_reports/wf/6wfs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  21653MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 | x 60
|
| 2 |
|
| 3 | x 5
|
| 4 | x 6
|
| 5 | 
|
| Symmetry | Point symmetry: (Schoenflies symbol: I (icosahedral)) |
-Components
| #1: Protein | Mass: 16791.104 Da / Num. of mol.: 4 / Fragment: UNP residues 1-149 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Hepatitis B virus genotype D subtype adwProduction host:  #2: Chemical | ChemComp-U0A / 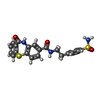  Mass: 453.534 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C22H19N3O4S2 / Feature type: SUBJECT OF INVESTIGATION Mass: 453.534 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C22H19N3O4S2 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Hepatitis B virus genotype D subtype adw / Type: VIRUS / Entity ID: #1 / Source: RECOMBINANT | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Source (natural) | Organism: Hepatitis B virus genotype D subtype adw / Strain: isolate United Kingdom/adyw/1979 | |||||||||||||||
| Source (recombinant) | Organism: | |||||||||||||||
| Details of virus | Empty: YES / Enveloped: NO / Isolate: OTHER / Type: VIRUS-LIKE PARTICLE | |||||||||||||||
| Buffer solution | pH: 7.5 | |||||||||||||||
| Buffer component |
| |||||||||||||||
| Specimen | Conc.: 10 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | |||||||||||||||
| Specimen support | Details: unspecified | |||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK III / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 295.15 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD |
| Image recording | Electron dose: 33 e/Å2 / Detector mode: SUPER-RESOLUTION / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 679 |
- Processing
Processing
| EM software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | |||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 24823 | |||||||||||||||||||||||||
| Symmetry | Point symmetry: I (icosahedral) | |||||||||||||||||||||||||
| 3D reconstruction | Resolution: 4.6 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 11080 / Symmetry type: POINT | |||||||||||||||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT / Space: REAL / Target criteria: Correlation Coefficient | |||||||||||||||||||||||||
| Atomic model building | PDB-ID: 6BVF Pdb chain-ID: A / Accession code: 6BVF / Pdb chain residue range: 1-143 / Source name: PDB / Type: experimental model |

